

不同检测中心的纳米粒子表面蛋白冠检测差异性分析——重复性分析
Gharibi, H., Ashkarran, A.A., Jafari, M. et al. A uniform data processing pipeline enables harmonized nanoparticle protein corona analysis across proteomics core facilities. Nat Commun 15, 342 (2024). https://doi.org/10.1038/s41467-023-44678-x
摘要 蛋白冠是在生物体液中的纳米颗粒上动态形成的一层主要由蛋白质组成的生物分子层,对于预测纳米药物的安全性和有效性至关重要。蛋白冠的蛋白质组成通常使用液相色谱-质谱法 (LC-MS/MS) 进行分析。我们最近的研究涉及由 17 个蛋白质组学机构分析的相同样本,强调了显著的数据变异性,在这些中心中只有 1.8% 的蛋白质被一致鉴定出来。在这里,我们实施了一个聚合数据库搜索,统一了可变修饰、酶特异性、允许的漏诊切割次数以及蛋白质和肽水平上严格的 1% 错误发现率等参数。这种统一的搜索极大地协调了蛋白质组学数据,提高了可重复性和不同核心中一致性鉴定的独特蛋白质的百分比。具体来说,在 717 种定量蛋白质中,253 种 (35.3%) 在前 5 家检测中心共享(16.2% 在前 11 家检测中共享)。此外,我们注意到还原和烷基化是蛋白冠样品处理中的重要步骤,正如预期的那样,省略这些步骤可将总定量肽的数量减少约 20%。这些发现强调了蛋白冠分析中标准化程序的必要性,这对于推进纳米级生物技术的临床应用至关重要。
简介
纳米颗粒一旦暴露于生物体液中,其表面就会迅速被一层离子和各种类型的生物分子的层覆盖,称为生物分子/蛋白冠。蛋白冠的组成,就参与蛋白质的类型、丰度和修饰而言,决定了生物系统(例如细胞)如何感知纳米颗粒并对其做出反应。最近对蛋白冠领域 2,134 篇已发表的论文进行的聚类分析揭示了纳米颗粒蛋白冠的蛋白质组学分析存在很大异质性(例如,就已鉴定的独特蛋白质的数量而言);因此,非常需要开发标准化方法/方案,以改善各种实验室/核心中纳米颗粒蛋白电晕的蛋白质组学表征。基于质谱的蛋白质组学通常会产生可重复的数据(reproducible data),在不同实验室进行的类似实验之间的主要区别在于给定样品中定量的蛋白质数量(即蛋白质组覆盖率)。因此,在蛋白质组覆盖率低的情况下,缺乏对低丰度真实靶标的检测可能会产生偏倚,因此可能会选择不太重要的靶标候选者进行后续分析。生物体液(例如血浆/血清)的蛋白质组学分析以及(血浆/血清)蛋白冠的分析都说明了这个问题,因为蛋白冠层中蛋白质的存在与否会导致数据误解或缺少生物标志物。蛋白冠的分析面临与血浆蛋白质组学类似的挑战,主要包括宽动态范围,即 22 种蛋白质占血浆蛋白质重量的 99%,来自此类蛋白质的肽挤满了质谱,阻碍了蛋白质组的深入分析,特别是对于丰度相当低的蛋白质。此外,另一个分析难点是血浆中存在不同的蛋白质亚型。事实上,已经开发了使用纳米颗粒蛋白冠的新兴技术,用于降低生物标志物发现中给定生物体液的生物学复杂性。尽管存在这些挑战,科学家们还是定量了血浆中数千种蛋白质,从而发现了不同的基于疾病的生物标志物。
虽然血浆蛋白质组学在不断改进,但在样品制备以及数据提取、清洁和处理方面,不同蛋白质组学研究的标准化尝试有限。然而,已经讨论了有关研究设计、血浆样品采集、质量保证、样品制备、MS 数据采集、数据处理和生物信息学以及蛋白质组学实验的最低报告要求的注意事项和建议。
给定核心测试机构报告的蛋白冠的质量和蛋白质组覆盖率可能会受到样品制备方案、分析柱、液相色谱 (LC) 系统和质谱 (MS) 仪器以及方法参数和分析持续时间的影响。其他变异来源包括原始文件的数据库搜索平台、搜索设置、错误发现率 (FDR) 的控制、翻译后修饰的包含以及使用的序列数据库。
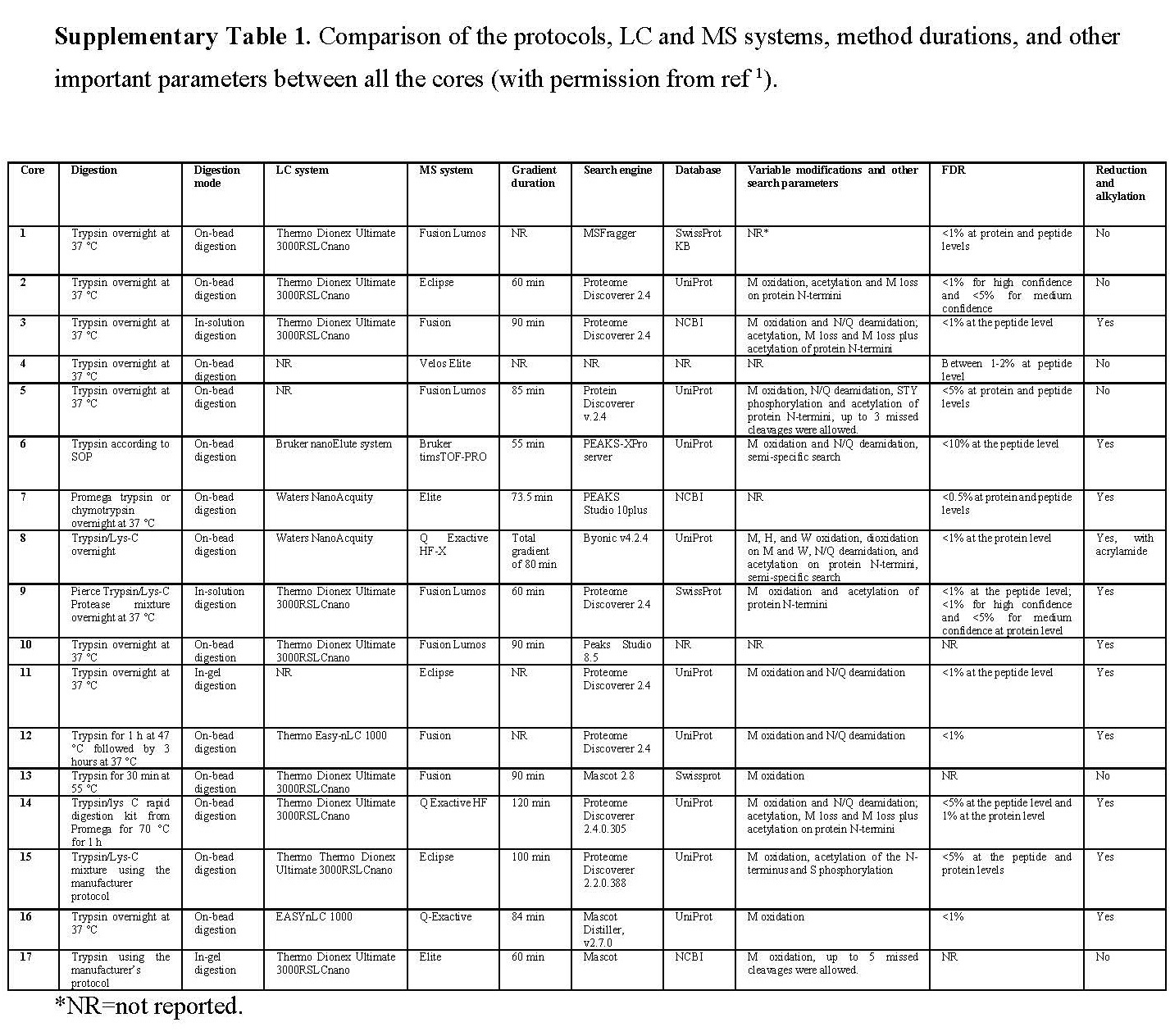
为了研究从不同测试中心获得的蛋白冠数据的异质性程度和来源,我们将相同的蛋白冠样品发送到不同的液相色谱-质谱 (LC-MS/MS) 核心测试设施,并分析其报告的结果。更具体地说,哈佛大学、斯坦福大学、麻省理工学院 (MIT)、凯斯西储大学、韦恩大学、伊利诺伊大学、康奈尔大学、田纳西大学、内布拉斯加大学林肯分校 (UNL)、密苏里大学、辛辛那提大学、佛罗里达大学、堪萨斯大学医学核心 (KUMC) 的中心分析了 17 个相同的蛋白冠样品等分试样, 德克萨斯大学圣安东尼奥分校 (UTSA)、密歇根州立大学 (MSU)、加州大学圣地亚哥分校 (UCSD) 和内华达大学里诺分校 (UNR)。分析分 3 个技术重复进行。没有提供或要求标准操作程序;相反,要求核心设施按照通常的做法分析样品。此后,我们用随机数(与之前的研究4 中的数字相同)对核心设施名称进行盲法,以防止任何潜在的利益冲突。实验方案的基本详细信息可在补充表 1 中找到。我们已经向所有中心索取了详细的方案,这些方案可以在我们原始研究4 的补充信息中找到。
在这项研究中,我们探讨了数据库搜索、数据提取、处理和分析对观察到的数据异质性的影响。具体来说,我们研究了采用具有统一参数(包括受控 FDR)的聚合数据库搜索是否有助于标准化和协调结果。
结果
我们对来自 15 个使用 Orbitrap 检测器的中心的 LC-MS/MS 原始文件进行了统一的数据库搜索(整个工作流程如图 2 所示)。中心 6 被排除在搜索之外,因为样品是通过 Bruker timsTOF-PRO 仪器分析的。中心 8 也被排除在外,因为丙烯酰胺用于半胱氨酸 (Cys) 残基的烷基化,而其他中心使用碘乙酰胺 (IAA) 或跳过烷基化步骤。由于几个中心没有说明是否进行了蛋白质的还原和烷基化,我们将 Cys 氨基甲基化作为可变修饰,并在蛋白质定量中使用氨基甲基化肽。虽然一些中心在其单独的数据库搜索中包括了其他修饰,例如脱酰胺化和磷酸化,但我们只将蛋白质 N 末端的常规蛋氨酸氧化和乙酰化作为可变修饰,因此结果在中心之间具有可比性。在蛋白质和肽水平上应用 1% 的 FDR,类似于以前的研究。如补充表 1 所示,不同的中心在蛋白质和/或肽水平上使用 1-10% 的 FDR,而一些中心没有说明 FDR 信息。以前一些中心使用半特异性检索,而这里我们只检索特异性胰蛋白酶肽。我们也最多只允许两次漏诊,这是相当标准的,并且在社区中被广泛接受。而以前几个中心在他们的个人搜索中允许多达 3-5 个遗漏的卵裂。值得注意的是,我们只强调参数中的这些变化是异质性的来源,并且只应用社区广泛接受的参数(例如,不超过 1% 的 FDR。这不会破坏由不同核心测试中心单独执行的先前数据库搜索的有效性。整个工作流程如图 1 所示。

统一数据库搜索显著提高了不同测试中心数据的均一性
去除污染物后,统一数据库搜索确定了 1,335 种蛋白质(补充数据 1)。与此同时,从不同核心设施中单独搜索的数据集的汇编导致鉴定了 4022 种蛋白质。我们认为,在之前的研究中鉴定出的蛋白质数量明显增加,部分原因是缺乏在蛋白质和肽水平上应用严格的 FDR,以及使用不同的搜索引擎、序列数据库和其他搜索参数,例如可变的翻译后修饰、酶特异性和允许的遗漏切割的数量。每个核心设施定量的蛋白质数量如图 2 所示。2a 并与之前研究中检测到的蛋白质进行了比较。正如预期的那样,由于统一搜索,所有中心的蛋白质数量都减少了。与我们的统一搜索相比,应用不太严格的 FDR、半特异性数据库搜索、包含具有两个以上缺失切割的肽以及包含非常规修饰(如磷酸化)预计将产生更多的蛋白质。因此,与单个数据库搜索中检测到的蛋白质数量相比,在统一搜索中,使用上述四个参数执行数据库搜索的中心定量蛋白质的数量减少了更多。
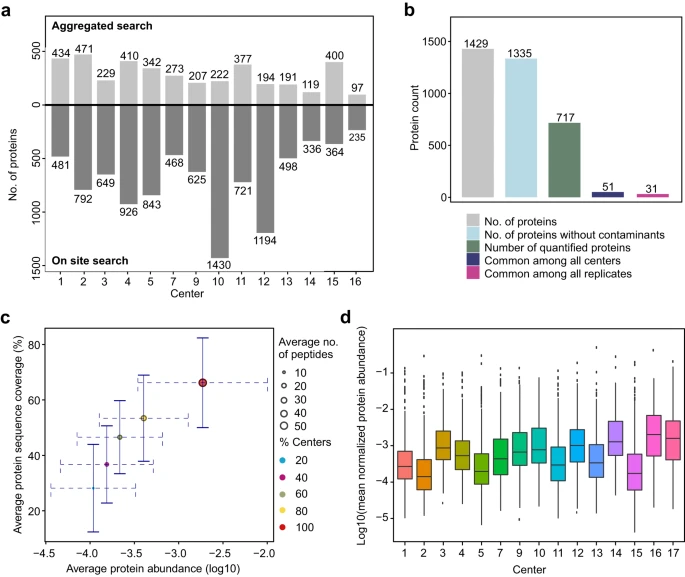
总体而言,定量了 717 种蛋白质,其中 51 种 (7.1%) 在所有中心之间共享(补充数据 2),在所有重复中发现了 31 种 (4.3%) 蛋白质(补充数据 2 中蛋白质的NA_count_all_replicates为零)(图 2)。在之前的研究中,只有 1.8% 的蛋白质组在 12 个核心设施之间共享。正如预期的那样,所有中心之间共享的大多数蛋白质具有相对较高的丰度、更高的检测到的肽数量和更高的序列覆盖率(图 D)。2c;例如,红色和黄色圆圈表示 100% 和 80% 的核心设施定量的蛋白质)。15 个核心的蛋白质水平强度分布如图 1 所示。我们还将所有核心设施中的 51 种共享蛋白质映射到 KEGG 通路,富集的通路如图 2 所示。
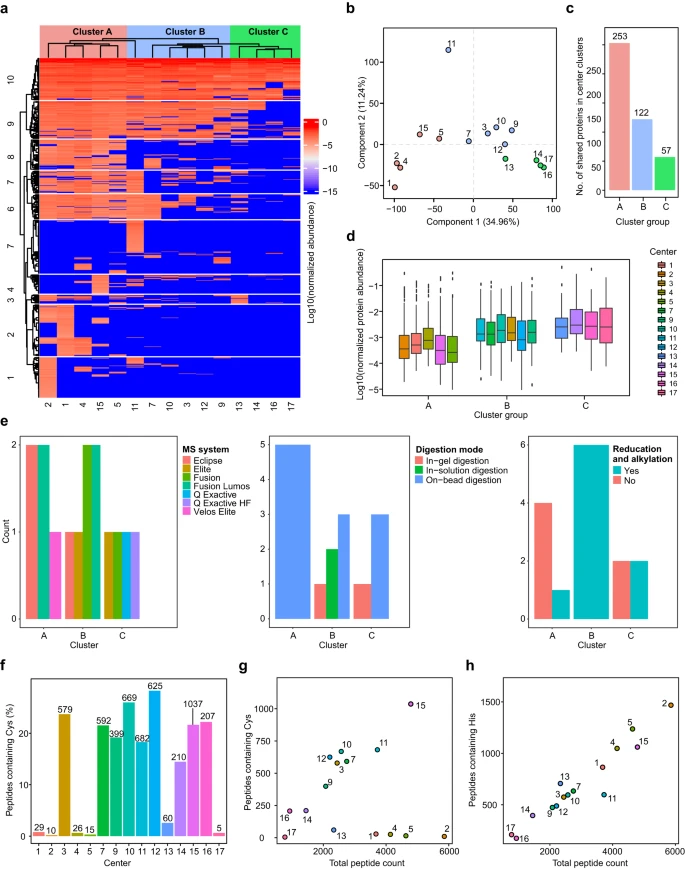
a 在 15 个核心设施的相同纳米颗粒蛋白电晕中定量的 717 种蛋白质的归一化强度的分层聚类。b 由 15 个核心设施定量的蛋白质组的 PCA 分析。c 在数据集的分层聚类和 PCA 分析中确定的不同簇中共享蛋白质的数量。面板 a-c 中的红色、蓝色和绿色是指中心簇。d 蛋白质强度在不同簇中的分布。箱线图:中心线,中位数;箱子限制包含 50%;上四分位数和下四分位数,75% 和 25%;maximum, 最大值(不包括异常值);最小值,最小值,不包括异常值;异常值,是上四分位数和下四分位数的 1.5 倍以上。e 在面板 a 中观察到的不同参数对聚类的影响。f 从每个核心设施获得的数据中至少包含一个 Cys 残基的肽的百分比(每个条形顶部的数字表示含 Cys 的肽的实际数量)。g 含 Cys 的肽数与总肽数的关系。h 含有 His 残基的肽数与肽总数的关系。所有分析均基于平均 3 个技术重复。
共享蛋白质在每个簇的核心设施之间的蛋白质水平强度分布如图 2 所示。4d,表明簇 C 的核心设施主要定量了样品中最丰富的蛋白质(簇 C 中共享蛋白质的平均蛋白质强度高于 B,反过来,B 的平均蛋白质强度高于簇 A)。这些结果表明,某些核心设施在定量低丰度蛋白质和达到更高的蛋白质组覆盖率方面优于其他设施。与我们之前的研究相比,这些发现表明,统一的数据库搜索极大地增加了不同核心设施之间共享蛋白质的数量,并且可以以无偏倚的方式比较每个中心的性能。假设,如果应用其他统一搜索参数,共享蛋白质也会增加。
为了进一步检查可能导致图 1 中观察到的聚类的其他因素。4a 中,我们调查了每个集群中内核使用参数的次数。如图 1 所示。4e 中,所用 MS 系统、消解模式以及包含还原和烷基化步骤方面存在聚类趋势。总体而言,高端(最近推出的)质谱仪在提供最高的蛋白质组覆盖率方面表现出色,但也有一些例外。意料之中,由于样品损失较低,磁珠上消化(尽管并非在所有情况下)总体上提供了比溶液内或凝胶内消化更高的蛋白质组覆盖率,这可能解释了为什么更多的核心机构根据他们的经验选择磁珠上消化。我们还研究了其他参数(包括梯度持续时间、变量修饰、液相色谱系统和 FDR)对图 1 中聚类的影响。4a. 但是,这些参数都没有对聚类产生明显影响。Cys 残基占蛋白质组中氨基酸的 2.3%,因此,≈20% 的所有胰蛋白酶肽至少包含一个 Cys21。在没有还原和烷基化的情况下,这些肽在消化过程中或消化后交联,并且大多数在清洁过程中被消除和/或通过常规数据库搜索未找到。因此,这些肽无法通过常规 LC-MS/MS 分析进行鉴定/定量。由于 6 个核心设施在提供的方案中未显示 Cys 还原和烷基化(中心 1、2、4、5、13 和 17),我们还计算了不同中心的定量含 Cys 肽的数量。如图 1 所示。4f 中,在其方案中包括还原和烷基化步骤的中心围绕预期的含 ~20% Cys 的肽进行定量。另一方面,上述 6 个中心未能量化此类肽。预计含 Cys 的肽数与总肽计数之间具有良好的相关性。在执行 Cys 残基还原和烷基化的中心中观察到这种相关性,但在跳过此步骤的 6 个中心中未观察到这种相关性(图 D)。作为对照,我们绘制了含组氨酸 (His) 的肽的数量与肽总计数的关系,证明了近乎完美的相关性(图 D)。4h)。我们选择了 His,因为 His 在蛋白质组中的频率非常接近 Cys 残基21。有趣的是,跳过还原和烷基化步骤的 4 个中心属于图 A 中的簇 A。4a 中,它量化了最多的蛋白质。然而,即使在这些情况下,数据质量(每个蛋白质的肽数和序列覆盖率)以及蛋白质组覆盖率也可以通过纳入含 Cys 的肽来进一步提高。总的来说,这一发现表明,在LC-MS/MS工作流程中包括Cys还原和烷基化步骤以防止数据丢失是必要的。
不同检测中心蛋白质组学数据的全局相似性
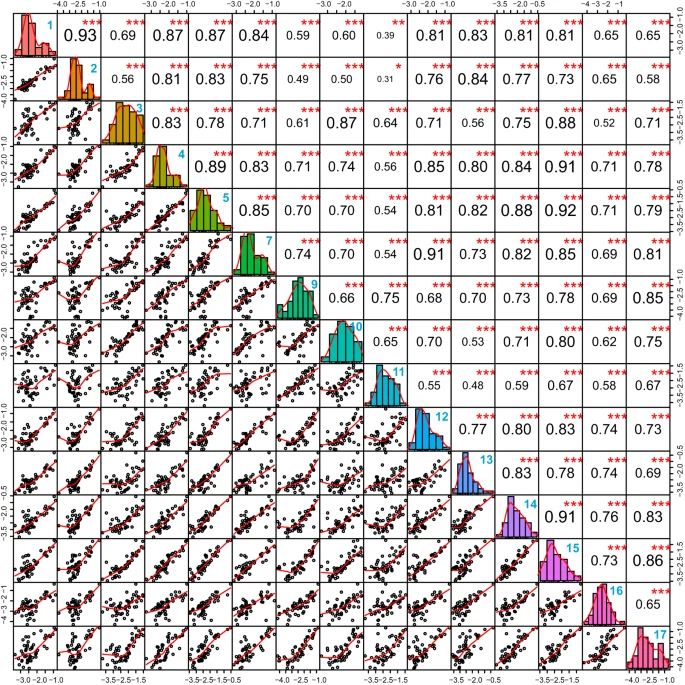
然后,我们计算了 15 个中心共享的 51 种蛋白质之间的相关性(图 D)。5). 这种无偏分析表明,对于一致定量的蛋白质,所有核心都报告了高度可比的数据。大多数核心设施的数据通常显示与其他设施的相关系数高于 0.7。只有来自 11 个核心的数据通常与其他设施的数据显示出低于 0.7 的相关性,我们无法将其归因于单个参数,例如消解时间、仪器类型、数据库搜索等。可接受的相关水平表明,来自每个核心的数据可以在给定的蛋白质组覆盖度内进行验证,并且数据的可变性主要源于不同的蛋白质组覆盖度。虽然LC-MS/MS方案和工作流程也可能产生差异,但本文表明,在蛋白质和肽水平上执行统一和标准的数据库搜索并严格控制FDR,可以显著协调不同核心设施的数据。
讨论
本研究的结果表明,使用统一且更标准的数据库搜索有助于在不同核心设施中均匀化蛋白冠分析的结果。跨中心检测到的蛋白质从 4,022 个减少到 1,335 个(定量)蛋白质,这表明数据库搜索参数设置对搜索结果有显著影响。统一的数据处理和分析增加了不同中心之间共享蛋白质的数量,并通过统一序列数据库、搜索参数和数据清理来提高数据可重复性。我们还表明,虽然共享蛋白质的丰度在不同中心之间通常具有良好的相关性,但变异性的主要来源来自不同程度的蛋白质组覆盖。在这种情况下,蛋白质组覆盖率在很大程度上取决于 LC-MS/MS 方案、工作流程、LC 和 MS 仪器、数据提取和处理。我们进一步证明,还原和烷基化是样品制备中必不可少的步骤,跳过此步骤会导致肽水平数据丢失 20%。缺乏对含 Cys 肽的检测会影响蛋白质序列覆盖率和每种蛋白质肽数方面的蛋白冠分析质量。我们进一步证实,提供较低蛋白质组覆盖率的中心主要量化了蛋白冠中更丰富的蛋白质。蛋白质组覆盖度在蛋白冠研究中非常重要,因为每种蛋白质的存在与否最终都会影响生物标志物的选择。分析表明,LC-MS/MS方案、工作流程、LC和MS仪器、数据提取和处理中的多个变量可以解释给定中心优于其他中心的原因。MS 仪器、消化模式以及包含还原和烷基化步骤是当前研究中影响蛋白质组覆盖率的变量之一。然而,有时,即使是几个跳过 Cys 还原和烷基化步骤的中心也优于执行这些步骤的其他中心。蛋白质组覆盖率似乎在很大程度上取决于工作流程、科学家以及总体上执行实验的中心。这强调了 LC-MS/MS 样品制备中良好实践的重要性,并强调了用户专业知识作为另一个有影响力的参数的重要性,正如之前所强调的那样。
有几项研究侧重于技术和工具方面,这些方面可以在从不同中心检索的数据中引入异质性,包括其他高通量技术,例如 RNA 测序和微阵列(尽管不是纳米颗粒蛋白冠)。其他研究调查了分析前样品处理和储存对血浆蛋白质组学的影响。我们已经在之前的报告中讨论了几项研究。
数据库搜索中的某些参数可能会对数据输出产生中度或显著影响。我们认为,搜索引擎(软件)的选择、使用的序列数据库、包含几个变量修改以及分配特定或半特异性搜索可以适度地影响数据输出,而 FDR 率可能会产生最大的影响。虽然不同的搜索引擎可能会产生略有不同的输出,但一旦它们经过测试,它们就会定期更新,并且它们的输出是可信的。包含其他修饰或进行半特异性搜索会增加定量肽的数量(因此,蛋白质的数量),但代价是假阳性发现率较高。选择较高的 FDR 临界值通常会导致鉴定出更多的蛋白质,但与此同时,假阳性命中的数量也会增加。肽水平上较高的 FDR 将允许大量错误识别的肽,这可能会在以后损害基于肽水平鉴定的蛋白质水平推断38。高 FDR 对于由少数肽检测到的低丰度蛋白质尤其有害。因此,这里我们将主要讨论 FDR 控制的重要性。
一些研究调查了 FDR 控制对不同中心蛋白质组学分析的影响。例如,Collins 等人5 对全球 11 个地点的所有理论碎片离子谱 (SWATH) MS 数据采集进行了顺序窗口采集的比较重现性分析。将一组连续稀释的标准肽加标到 HEK293 细胞裂解物中。虽然所有站点都使用了 SCIEX TripleTOF 5600/5600+ 质谱仪,但纳离子色谱系统具有不同的型号,但来自同一供应商 SCIEX。该研究检测到一组核心 4,077 种蛋白质,这些蛋白质在 >80% 的样品中被一致检测到。与我们研究的结果类似,当数据分析和 FDR 控制在逐个站点的基础上独立进行时,注意到站点之间一致检测到的蛋白质减少。但是,应该注意的是,没有进行样品制备,因此工作流程的变化仍然很小。上述示例表明,通过集中样品制备和数据分析,以及最大限度地减少仪器参数的变化,可以实现复杂样品中大多数蛋白质的重现性。
与上述研究相比,相同的蛋白冠样品被运送到不同的中心,随后的样品制备和 LC-MS/MS 分析由中心自行决定进行,没有要求/提供标准化的仪器参数或方案。因此,当前研究中唯一的汇总步骤是蛋白质样品的制备、数据提取、处理和分析。此外,我们研究中的中心必须执行数据依赖性采集 (DDA)。与 SWATH 不同,DDA 采集涉及随机碎片离子 (MS2) 采样,因此肽采样的可重复性较低; 即,当母离子的数量超过母离子选择周期的数量时,母离子选择变为随机。
人类蛋白质组组织 (HuPO) 测试样品工作组将 20 种高度纯化的重组蛋白的等摩尔混合物分发给 27 个不同的实验室,这些实验室依次根据自己的常规和方案分析样品,没有任何限制。在 27 个实验室中,只有 7 个实验室正确报告了所有 20 种蛋白质。对原始数据的集中分析表明,在所有 20 个实验室中都检测到了所有 27 种蛋白质。漏诊鉴定或假阴性、环境污染、数据库匹配问题以及蛋白质鉴定的管理被发现是异质性的来源。研究表明,在肽鉴定和蛋白质分配中观察到的主要变异性是由数据处理和分析的差异引起的,而不是由数据收集的差异引起的。
在这里,我们证明,通过在蛋白质和肽水平上应用严格的 FDR 临界值并统一其他搜索参数,例如酶特异性、可变翻译后修饰和考虑遗漏切割的肽,与在每个位点进行单独数据库搜索相比,一致定量蛋白质的百分比增加了 9 倍。与我们之前的研究相比,由不同的核心机构进行独立的数据库搜索,鉴定出 4022 种蛋白质,而我们在这里鉴定了 1429 种蛋白质,仅量化了 717 种蛋白质。
我们认为,与其他类型的蛋白质组学分析(例如细胞和组织)相比,血浆蛋白质组学通常具有挑战性,这是蛋白冠层中一致检测到蛋白质的比率较低的主要原因之一。同样,在之前的研究中,作者重新分析了 2005-2017 年的 178 项实验数据,表明只有 50% 的研究报告了 500 种最丰富的血浆蛋白。
总之,我们揭示了使用统一的数据库搜索为使用 LC-MS/MS 在蛋白冠研究中采取措施和质量控制提供了机会。这种方法为协调蛋白冠结果的数据分析铺平了道路,使利益相关者能够对现有文献中的蛋白质组学数据进行荟萃分析。它旨在最大限度地减少由于不同实验室的样品制备和工作流程差异而引起的冲突和差异。提高蛋白冠研究中的可重复性和蛋白质组覆盖率可以加速纳米医学技术在诊断和治疗应用中的成功临床转化。
PuriMag™蛋白冠前处理磁珠试剂盒|高深度蛋白质组学(http://www.purimagbead.com/Product/8096211554.html)
- 上一篇:质谱指纹图谱专家共识 | 蛋白/多肽指纹图谱在肿瘤中的临床应 2025/1/7
- 下一篇:血液蛋白质组的检测深度——如何看待不同的策略方案? 2024/12/22