
使用 SP3 纯化基于凝胶的样品分级用于自上而下的蛋白质组学
Gel-Based Sample Fractionation with SP3-Purification for Top-Down Proteomics
Ayako Takemori, Naoyuki Sugiyama, Jake T. Kline, Luca Fornelli, and Nobuaki Takemori
Journal of Proteome Research 2025 24 (2), 850-860
DOI: 10.1021/acs.jproteome.4c00941
摘要:蛋白质组样品的精确预分馏是在自上而下的蛋白质组学中实现深入分析的有效方法。PEPPI-MS(被动洗脱聚丙烯酰胺凝胶中蛋白质作为 MS 的完整物质)是一种基于凝胶的样品分级分离方法,通过在 SDS-PAGE 分离后从聚丙烯酰胺凝胶中高效提取蛋白质,实现基于分子量的高分辨率蛋白质组分离。此后,在质谱分析之前,必须有效地去除 PEPPI 馏分中的 CBB 和 SDS 等污染物。在这项研究中,通过将 PEPPI-MS 与 SP3(单罐固相增强样品制备)中使用的基于磁珠的蛋白质纯化方法相结合,我们开发了一种完整、稳健且简单的样品制备工作流程,用于自上而下的蛋白质组学在 PEPPI-SP3 中,从凝胶中提取的蛋白质收集在 SP3 珠子的表面,用有机溶剂洗涤,并用含有 0.05% (w/v) SDS 的 100 mM 碳酸氢铵完整回收。使用阴离子交换 StageTip 进行额外纯化后,对回收的蛋白质进行质谱分析。使用人细胞裂解物进行的性能验证显示,与使用有机溶剂沉淀或超滤的常规 PEPPI 工作流程相比,低分子量蛋白质回收率显著提高,变异系数更低。
SP3磁珠请参考 http://www.purimagbead.com/Product/1736805216.html
介绍
单个基因在体内产生具有不同化学结构的各种翻译产物,称为蛋白质形式。蛋白形式有助于蛋白质相互作用和亚细胞定位的多样性,从而扩展蛋白质的生理功能。虽然蛋白质形式的总数仍然未知,但据估计,该数量比人类基因的数量22,800 个高出 100 多万个 。了解庞大的人类蛋白质组学是当今蛋白质组学研究的目标之一,为实现这一目标,迫切需要开发全面的蛋白质组分析技术。
自下而上的蛋白质组学 (Bottom-up proteomics, BUP) 是人类蛋白质组大规模分析的主要分析方法,用于分析通过酶消化获得的肽片段,原则上通常难以应用于蛋白质形式的高度准确鉴定。因此,允许直接分析完整蛋白质形式的自上而下的蛋白质组学 (Top-down proteomics, TDP) 被用作用于此目的的主要分析方法。在 TDP 分析中,样品制备的影响不容小觑,正如 Kaulich 等人最近的一份报告中所详细描述的那样。蛋白质形态鉴定通常是通过反相液相色谱 (LC) 或毛细管电泳 (CE) 分离完整的蛋白质形态,然后在线连接质谱仪中碎裂来实现的。然而,为了从生物样品中检测出更多的蛋白质组分,在 LC/CE 分离之前进行样品预分馏是必不可少的。
SDS-聚丙烯酰胺凝胶电泳 (PAGE) 是生化实验中的一种核心蛋白质分离方法,通过在聚丙烯酰胺凝胶中添加阴离子表面活性剂 SDS,根据分子量 (MW) 对线性化蛋白质进行电泳分离。SDS-PAGE 的特性能够对细胞裂解物中的复杂蛋白质组进行高分辨率分离,并被广泛用作 BUP 中一种强大的样品预分馏方法。2020 年,我们开发了 PEPPI-MS,这是一种用于被动提取凝胶中蛋白质的高效方法,并成功地显著提高了凝胶中蛋白质的回收率。尽管被动提取通常简单且具有成本效益,但它也与低回收率、长提取时间和难以应用于高 MW 蛋白质有关。PEPPI-MS 通过使用考马斯亮蓝 (CBB) 和 SDS 作为提取增强剂解决了这些缺点,即使对于高 MW 蛋白,也能在 10 分钟内实现高回收率。
在 PEPPI-MS 中,蛋白质组按如下方式分级分离:(1) 通过 SDS-PAGE 分离后在凝胶中添加 CBB,(2) 切除感兴趣 MW 区域的样品泳道,(3) 将凝胶块捣碎并在含有 0.05–0.1% (w/v) SDS 的 100 mM 碳酸氢铵溶液 (pH 8) 中被动萃取 10 分钟。由于获得的馏分含有 CBB 和 SDS, 这会干扰 MS 分析,因此蛋白质纯化是必不可少的,目前使用甲醇-氯仿-水沉淀 (MCW) 或阴离子交换转盘辅助顺序样品制备 (AnExSP)。MCW 适用于约 300 μL 的小体积溶液,例如 PEPPI 馏分,是一种廉价且简单的蛋白质纯化方法。AnExSP 是一种使用阴离子交换固相萃取 (SPE) 微量离心柱(称为 AX-StageTip)从样品中去除 SDS 和 CBB 的方法。AnExSP 允许纯化 PEPPI 组分,而不会损失低 MW 蛋白,这通常是 MCW 的一个问题。在 AnExSP 中,CBB、SDS 和 PEPPI 馏分中的蛋白质最初被捕获在 StageTip 的阴离子交换 SPE 盘上,在随后的洗脱过程中,使用乙醇和/或含有甲酸 (FA) 的乙腈溶液回收蛋白质。然而,标准 300 μL PEPPI 馏分中的 SDS 量超过了 AX-StageTip 可能去除的限度,需要事先进行 FASP 处理,这是一种使用离心超滤装置进行繁琐的尿素清洗。
在本研究中,我们尝试开发一种使用 SP3 进行 PEPPI 分级分离的新纯化方法,SP3 是目前 BUP 的主流样品预处理方法。 在 SP3 中,通过添加有机溶剂(如乙醇或乙腈)沉淀的蛋白质被吸附到羧酸盐修饰的磁珠表面,回收的蛋白质直接在磁珠上进行酶消化。通过可靠、简单的作选择性回收蛋白质,可有效去除 SDS 和其他污染物,其特点是在处理痕量蛋白质样品时具有很高的重现性。结合最近开发的用于多样品高通量处理的商业自动化系统,该方法可应用于多样品处理。SP3磁珠请参考 http://www.purimagbead.com/Product/1736805216.html
尽管 SP3 具有出色的样品纯化特性,但目前其在 TDP 中的应用受到限制,部分原因是难以回收吸附在磁珠上的完整蛋白质。Webb 小组的一项开创性研究报告称,在 −80 °C 下用 80% (v/v) FA 孵育可有效从磁珠中回收完整蛋白质,但条件对于高通量应用来说太苛刻了。 为了使 SP3 适应 TDP,我们开发了新的实验条件,允许在室温下从磁珠中快速且可重现地回收蛋白质,从而为TDP 提供了一种简化而高效的样品分离工作流程,该工作流程结合了基于 PEPPI 的蛋白质组分分离和基于磁珠的馏分纯化,称为 PEPPI-SP3。PEPPI-SP3 实现的基于 MW 的高分辨率蛋白质组分离可最大限度地减少低 MW 蛋白的损失,从而实现高通量样品制备、全面分析,并最终实现多样品处理。
实验部分
补充方案中给出了先前发布的程序(如样品制备、SDS-PAGE、PEPPI 分级分离和蛋白质纯化)的详细方案。
材料
除非另有说明,否则用于 PEPPI 分级分离和蛋白质纯化的试剂购自 Wako(日本大阪),用于 TDP 分析的试剂购自 Fisher Scientific(美国伊利诺伊州罗克福德)。
细胞样品
本研究使用了 MS 相容的人蛋白提取物 (Promega, Madison, WI, USA),一种市售的人类细胞蛋白提取物 (HCPE)。在 SDS-PAGE 之前,通过将 100 μL HCPE(1 mg 蛋白质)与 1 μL 500 mM 二硫苏糖醇 (DTT) 在 37 °C 下孵育 90 分钟进行还原处理,然后通过在 23 °C 下与 1 μL 1 M 碘乙酰胺在黑暗中孵育 30 分钟进行烷基化处理。在 Amicon 离心式 3 kDa 超滤装置(Merck Millipore,达姆施塔特,德国)中用 0.05% (w/v) SDS/100 mM 碳酸氢铵 (ABC) 替换溶剂,并调节至 2 μg/μL 的蛋白质浓度。
SDS-PAGE
使用 4–12% 预制凝胶 NuPAGE bis-tris(1 mm 厚,10 孔)和 NuPAGE MES 电泳缓冲液(Thermo Fisher Scientific,Waltham,MA,USA)进行 SDS-PAGE。将样品与 NuPAGE LDS 上样缓冲液 (Thermo) 混合,然后上样到凝胶孔中。在 180 V 的恒定电压下进行电泳后,从凝胶盒中取出凝胶,用 EzStain AQua(ATTO,Tokyo,Japan)染色 8 分钟,然后用去离子水洗涤 30 分钟至 1 小时。对于 TDP,进行了以下调整:使用自制的 10% 凝胶(1 mm 厚,10 孔)代替 NuPAGE 4–12% 凝胶(1 mm 厚,10 孔),并使用 Bio-Safe 考马斯染色剂(Bio-Rad,Hercules,CA)染色 60 分钟,而不是 EzStain AQua 染色 8 分钟。
PEPPI 分级分离
使用 MW 标记作为指示剂,用工艺刀在感兴趣的 MW 区域对染色凝胶的样品泳道进行切片。将切除的凝胶块收集在 BioMasher II 管(Nippi,Tokyo,Japan)中,并用塑料杵精细研磨。将凝胶进一步与 250 μL 0.05% (w/v) SDS/100 mM ABC 混合,并在管式混合器中以 23 °C、1500 rpm 摇动 10 分钟。使用离心过滤器去除凝胶,所得溶液(约 250 μL),即 PEPPI 组分,用于后续的蛋白质纯化:MCW、FASP 或 SP3。
MCW
将 PEPPI 馏分转移至 1.5 mL 微管中,加入 600 μL 甲醇、150 μL 氯仿和 400 μL 超纯水,混合,并在 23 °C 下以 13,500 rpm 离心 3 min。 离心形成双层溶液,其中去除上层,向剩余的底层加入 400 μL 甲醇,并在 23 °C 下以 13,500 rpm 离心 3 分钟。 用 400 μL 甲醇洗涤所得蛋白质沉淀物,并风干 30 分钟。
FASP
将 PEPPI 馏分转移至 Amicon 离心式 3 kDa 超滤装置中,然后用 8 M 尿素置换溶剂,再用 100 mM ABC 置换溶剂。如前所述,通过 AnExSP(自制 AX-StageTip)纯化装置中的溶液:(11) 使用前用甲醇洗涤 AX-StageTip 并用 100 mM ABC 平衡;将样品上样至 AX-StageTip 上,用 40 μL 100 mM ABC 洗涤,用 40 μL 0.5% (v/v) FA/50% (v/v) 乙醇洗脱,再用 40 μL 0.5% (v/v) FA/50% (v/v) 乙腈洗脱,最后三个步骤分别以 7000 rcf 离心 3 分钟。在 LC-MS 分析之前,将所得洗脱液在离心蒸发器中干燥。
SP3
SP3磁珠请参考 http://www.purimagbead.com/Product/1736805216.html。将购自 Cytiva(美国马萨诸塞州马尔堡)的 Sera-Mag SpeedBead 羧酸盐修饰的 E7 磁珠(50 mg/mL,货号 45152105050250)和 E3 磁珠(50 mg/mL,货号 65152105050250)各 50 μL 添加到 1.5 mL 微管中。用 800 μL 超纯水洗涤 3 次后,将珠子悬浮在 250 μL 超纯水中并储存在 4 °C 直至使用。将先前制备的 PEPPI 组分转移至 1.5 mL 微管中,并与 25 μL SP3 珠悬浮液 (20 μg/μL) 和 1.2 mL 乙醇混合;在 23 °C 下以 1200 rpm 振荡 10 分钟后,将微管置于磁力架上 3 分钟,并通过抽吸除去液体。用 800 μL 80% (v/v) 乙醇洗涤磁珠两次,用 800 μL 乙腈洗涤一次。为了回收吸附在磁珠上的完整蛋白质,在 23 °C 下用 20 μL 0.05% (w/v) SDS/100 mM ABC 在微管中以 2000 rpm 振荡磁珠 5 分钟。 将微管置于磁力架上 3 分钟,并如上所述在 fasp 后半部分对含有回收蛋白质的溶液进行 AnExSP 纯化。
ContamSpot 检测
通过 ContamSpot 测定法测定纯化后 PEPPI 组分的残留 SDS 浓度。(19) 将纯化的 PEPPI 馏分溶于 10 μL 0.1% (v/v) FA/2% (v/v) 乙腈中,用于测定。将 2 μL 样品、2 μL 0.1% (w/v) 邻甲苯胺蓝和 5 μL 乙酸乙酯混合在 0.2 mL PCR 管中,并以 2000 rpm 离心 15 秒。从通过离心分离成两层的溶液中,将 1.5 μL 乙酸乙酯层点样到薄层色谱 (TLC) 板上。
胰蛋白酶/Lys-C 消化
在 LC-MS 分析之前,使用 RapiGest(沃特世,美国马萨诸塞州米尔福德)溶解 PEPPI 馏分,并使用 MS 级胰蛋白酶/赖氨酸-C 混合物 (Promega) 消解。将来自不同 4 个 MW 区域的纯化 PEPPI 组分溶于 10 μL 0.1% (w/v) RapiGest/100 mM ABC 中,并混合在 1.5 mL 微管中。将混合级分(总共 40 μL)与 0.2 μg 胰蛋白酶/赖s-C 混合物混合,并在 37 °C 下消化 16 小时。 为了降解 RapiGest,向消解的样品中加入 10 μL 2.5% (v/v) 三氟乙酸 (TFA),并在 37 °C 下孵育 30 分钟。 在 23 °C 下以 13,500 rcf 离心 10 分钟后,通过自制的 SDB-StageTip 纯化上清液并进行 LC-MS 分析。
消化肽的 NanoLC/MS/MS
在 timsTOF Pro2 质谱仪(Bruker Daltonics,Bremen,Germany)上使用 CaptiveSpray 纳离子喷雾源 (Bruker Daltonics) 结合 nanoElute 2 纳流 UHPLC 系统 (Bruker Daltonics) 获取 MS 和 MS/MS 数据。在 50 °C 下,使用 PepSep ULTRA C18(250 mm × 75 μm、1.5 μm,Bruker Daltonics)作为分析柱。 将干燥的肽混合物溶于 30 μL 0.1% (v/v) TFA 和 5% (v/v) 乙腈中,每次进样使用 2 μL 溶液进行 LC/MS/MS。流速为 400 nL/min,流动相由 (A) 0.1% (v/v) FA 的水溶液和 (B) 0.1% (v/v) FA 的乙腈溶液组成。采用多级线性梯度:流动相B在60 min内从4%增加至20%,在30 min内从20%增加至28%,在15 min内从28%增加至40%,在5 min内从40%增加至100%,并在100% B下保持10 min。
timsTOF 在 PASEF (parallel accumulation and serial fragmentation) 模式下运行。施加 1500 V 的毛细管电压、3.0 L/min 的干燥气体和 180 °C 的干燥温度。MS 和 MS/MS 扫描范围为 m/z 100–1700,1/K0 范围为 0.6 至 1.6 Vs/cm2,斜坡时间为 100 ms,累积时间为 100 ms。碰撞能量根据离子淌度线性递增,从 1/K0 = 1.60 Vs/cm2 的 59 eV 到 1/K0 = 0.60 Vs/cm2 的 20 eV。对于数据依赖性采集 (DDA) 分析,每个周期进行 1 次 MS 扫描和 10 次 PASEF MS/MS 扫描,预定目标强度为 20,000。使用多边形过滤器,根据 m/z 和离子淌度选择单电荷和低 m/z 离子,从源离子母离子选择中排除。主动排除时间设置为 0.4 min。对于数据非依赖型采集 (DIA) 分析,DIA 窗口设置如表 S1 所示。MS 原始数据和分析文件已通过 jPOST 合作伙伴存储库 (https://jpostdb.org) 存放到 ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org),数据集标识符为 PXD056413。
DDA 和 DIA 分析
使用 MSFragger (v. 4.1) FragPipe (v. 22.0) 处理 DDA 数据,以鉴定肽和蛋白质,并生成用于 DIA 数据分析的光谱库。根据人类 SwissProt 数据库(2024 年 4 月 16 日从 UniProtKB 下载,仅限规范,40,870 个条目,包含 20,435 个反向诱饵)搜索 MS/MS 谱图。将母离子和子离子的质量精度设置为 20 ppm,并启用质量校准和参数优化。将酶特异性设置为胰蛋白酶/P,允许最多一个缺失的切割位点。半胱氨酸的氨基甲酰甲基化被设置为固定修饰,蛋氨酸的氧化和蛋白质 N 末端的乙酰化被允许为可变修饰。使用 Philosopher 中的 PeptideProphet 和 ProteinProphet 过滤结果,FDR < 1%。用于 DIA 数据分析的光谱库是根据所有 DDA 数据的结果生成的。
对于 DIA 数据,通过 FragPipe (v. 22.0) 用 DIA-NN (v. 1.8.2 beta 8) 定量蛋白质,蛋白质水平 FDR < 1%。针对不同的运行分别确定质量精度和扫描窗口。定量结果使用 MetaboAnalyst (v. 6.0) 和 PlotsOfData 可视化。
自上而下的样品制备、PEPPI 分级分离和处理
在手铸的 10% SDS-PAGE 上分离 9 个 40 μg 的 HCPE 样品,切除对应于 0-30 kDa 的区域,并如上所述进行 PEPPI。如上所述,用 MCW 、 FASP 和 SP3 方法处理 3 组。请注意,在浓缩之前,将 AnExSP 处理的样品在液氮中快速冷冻。使用 SpeedVac 进行溶剂蒸发,不加热,直到样品体积减少至 ∼5 μL(约 60–120 分钟;因样品而异)。浓缩后,使用 0.1% (v/v) FA、4.9% (v/v) 乙腈的 LC-MS 级水溶液将样品体积定为 20 μL。
色谱和自上而下的质谱
通过纳米毛细管高性能液相色谱在线与 EasySpray 纳离子喷雾离子源(Thermo Fisher Scientific,加利福尼亚州圣何塞)联用,进一步分离重悬的 HCPE 馏分(FASP 和 SP3 样品进样体积为 2 μL,MCW 进样体积为 3.5 μL)。使用Ultimate 3000色谱系统(Thermo Fisher Scientific)进行反相液相色谱,将流动相B的梯度从5%提高到2 min从5%提高到14%,然后在50 min内从14%提高到42%,然后连续两次以85% B清洗色谱柱,持续1 min,最后以5% B再平衡相,持续8.5 min, 保持 1.5 μL/min 的流速。流动相A由4.9% (v/v)乙腈水溶液在0.1% (v/v) FA中存在而成,而流动相B由4.9% (v/v)水的乙腈溶液和0.1% (v/v) FA组成。所有流动相组分均为LC-MS纯度等级(Fisher Scientific)。将样品直接进样到带有集成发射器的 MAbPac EasySpray 色谱柱上(Thermo Fisher Scientific;粒径 4 μm,长度 15 cm,内径 150 μm),使用集成的 EasySpray 柱温箱加热至 55 °C。通过施加 2.1–2.2 kV 电位产生纳电喷雾。所有质谱测量均在配备 FAIMS Pro 装置的 Orbitrap Eclipse 三组质谱仪 (Thermo Fisher Scientific) 上进行。在“蛋白质模式”下进行自上而下的实验,离子路由多极压力为 3 mTorr。源区域参数包括加热传输毛细管的 320 °C 温度、30% RF 振幅和 15 V 源内碎裂,以促进脱溶剂和去除不稳定的加合物。对于 DDA,使用 5 × 105充电和 50 ms 的自动增益控制 (AGC) 目标,最大进样时间为 50 ms,最大进样时间为 50 ms,在 400–2000 m/z 窗口内以 120,000 分辨能力(200 m/z 时)以 120,000 m/z 的分辨率(即单次微扫描)鉴定母离子。选择四极杆(3 m/z 隔离窗口)进行 HCD MS2 碎裂(35% 归一化碰撞能量),在 5–50+ 的电荷态范围内,强度阈值为 2.5 × 104。在 Orbitrap 质量分析器中收集碎裂谱图,在 400–2000 m/z 窗口内,无光谱平均,AGC 目标为 2.5 × 105 次充电,最大进样时间为 500 ms。FAIMS 补偿电压 (CV) 在单次运行中变化,每个样品单独运行 3 次(每个样品总共 9 个不同的 CV), 与之前的报告类似;(29) 运行 1:−40、−20 和 0 V;运行 2:−60、−50 和 −30 V;运行 3:−10、5 和 15 V;每个 CV 使用 1.2 s 的循环时间。应用动态排除(持续时间 30 s)。所有质谱数据文件均已上传到 MassIVE(存储库编号 MSV000095992)
自上而下的数据分析
将来自所有三种处理方法的 RAW 文件与 ProSight PD v. 4.2 (Proteinaceous, Inc., Evanston, IL, USA) 一起搜索,在 Proteome Discoverer 3.0 环境 (Thermo Fisher Scientific) 中作为节点运行,对照人类数据库。使用提供的 High-High 处理和共识工作流程进行数据分析。对于 FAIMS 文件,Spectrum Selector (光谱选择器) 节点中的 FAIMS CV 设置未指定,以适应每个文件使用多个 CV。所有谱图都在 High-High cRAWler 节点内进行去卷积:使用 Xtract 利用滑动窗口算法从 MS1 谱图中去除母离子质量数,扫描偏移量为 1,合并容差为 30 ppm,同时要求在至少 3 个滑动窗口检测中检测至少 3 种电荷态。在共识工作流程中定量之前,特征组需要 2.2 Da 的耐受性。应用了两个数据库搜索:使用 2.2 Da 母离子质量容差的注释蛋白形式搜索,以及基于 15 ppm 母离子容差的子序列搜索。对于这两个检索,片段容差均设置为 10 ppm。由单个前体产生的蛋白形式数量限制为 1。从 ProSight PD 中鉴定出的蛋白质形式和 UniProt 登录号以 1% FDR 过滤(对蛋白质形式、同工型和 UniProt 登录水平进行三次单独的 FDR 计算)。完成数据库搜索后,针对每种处理方法过滤生成的 tdReport,并根据开发人员的建议重新计算 Q 值。对于水肿总平均值 (GRAVY) 和等电点 (pI) 分析,使用 GraphPad Prism 10 (GraphPad Software, Boston, MA, USA) 中的内置函数,使用 Kruskal-Wallis 和 Dunn 多重检验校正,根据每组的平均值进行统计检验。使用 DeepTMHMM 预测鉴定蛋白质的跨膜状态。绘图是使用 GraphPad Prism 10 生成的。
结果与讨论
通过 SP3 纯化 PEPPI 组分
我们尝试根据先前发布的 BUP15 方案,以 250 μL 的样品规模(典型的 PEPPI 馏分体积)建立 TDP 的 SP3 方案。使用的磁珠是两种不同羧酸盐修饰磁珠的等重混合物,即 Cytiva Sera-Mag SpeedBead 羧酸盐修饰磁珠的亲水和疏水版本。在 BUP 的 SP3 样品制备中,通常建议在蛋白质中添加 10 倍重量的磁珠。 为了应用于 TDP,我们从 10 μg HCPE 中生成了四种不同 MW 范围的 PEPPI 级分,并检查了当添加不同量的珠子时与珠子结合的蛋白质量(图 S1)。在 250 至 20 kDa 的 MW 范围内,无论添加的磁珠量如何,回收率都相似,但在低于 20 kDa 的范围内,当添加的磁珠量小于 500 μg 时,回收率略有下降。因此,我们在本研究中将珠子的量设置为 500 μg。
为了在室温 (23–25 °C) 下快速回收磁珠上紧密吸附的蛋白质,我们认为需要表面活性剂辅助,并选择使用 SDS,它可以被 AnExSP 去除。具体来说,我们将 20 μL 0.05% (w/v) SDS/100 mM ABC(之前已被证明适用于 AX-StageTip 纯化)与洗涤的珠子混合,并在室温下用试管振荡器摇动混合物(图 1A)。HCPE 分析表明,0.05% (w/v) SDS/100 mM ABC 可以在 10 分钟内从珠子中回收蛋白质(图 1B)。作为 SDS 的替代品,我们还研究了 RapiGest、尿素、NDSB-195 和辛基葡糖苷的使用,这些物质可以在 MS 分析前轻松去除。尿素、NDSB-195 和辛基葡萄糖苷在摇动 10 分钟后没有产生任何显着的蛋白质回收(图 S2),可酸降解表面活性剂 RapiGest 产生相当于 SDS,但随后的酸处理导致回收的蛋白质降解(图 S3)。因此,我们决定在本研究中继续使用 SDS 进行蛋白质回收。
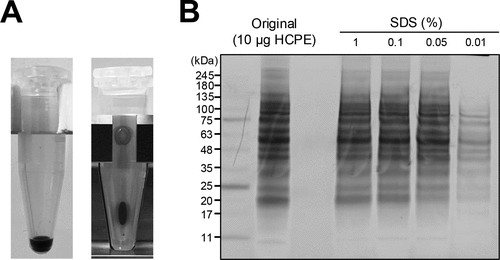
图 1.使用 SDS 溶液从 SP3 微珠中回收蛋白质。(A) 从 SP3 微珠中回收蛋白质。通过在 23 °C 下在 20 μL 0.05% (w/v) SDS/100 mM ABC 中摇动珠子 10 分钟(左图),回收吸附在 SP3 珠子 (500 μg) 上的 HCPE (10 μg)。蛋白质回收后,将试管放在磁力架上以去除磁珠(右)。(B) 从不同 SDS 浓度 (0.01–1% (w/v)) 的磁珠中回收的蛋白质的 SDS-PAGE 图像。条带用 CBB 染色。SP3磁珠请参考 http://www.purimagbead.com/Product/1736805216.html
PEPPI-SP3 工作流程
基于上述结果,我们建立了一个新的实验工作流程 PEPPI-SP3,通过在 AnExSP 纯化后结合基于 SP3 的蛋白质回收来纯化 PEPPI 组分(补充方案)。PEPPI-SP3 工作流程的方案如图 2 所示。在工作流程中,蛋白质组学样品通过 SDS-PAGE 分离,然后用 PEPPI 进行分级分离。将乙醇添加到 500 μg SeraMag 珠子和所得 PEPPI 级分中,以产生 80% (v/v) 乙醇溶液,并剧烈摇动以将沉淀的蛋白质成分吸附到珠子上(图 S4A 和 S4B)。使用磁力架分离珠子,并用 80% (v/v) 乙醇洗涤两次,用乙腈洗涤一次,以去除 CBB 和 SDS(图 S4C-S4E)。通过用 0.05% (w/v) SDS/100 mM ABC 摇动珠子 10 分钟,从珠子中回收蛋白质,并在 AnExSP 纯化后进行 LC-MS。
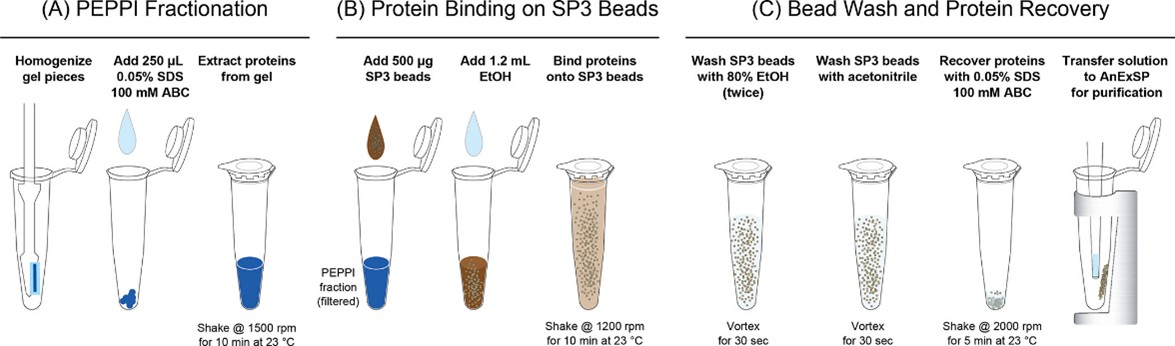
图 2.用于自上而下的蛋白质组学的 PEPPI-SP3 工作流程。(A) PEPPI 分级分离。样品 SDS-PAGE 分离后,用 CBB 对分离的蛋白质进行染色,并切除样品泳道中所需的分子量范围。使用一次性塑料捣碎管对切除的凝胶块进行匀浆,并用 0.05% (w/v) SDS/100 mM ABC 被动提取凝胶中的蛋白质。(B) 蛋白与 SP3 微珠的结合。将蛋白质回收溶液(PEPPI 组分)与 SP3 珠子和乙醇混合,搅拌,沉淀的蛋白质吸附到珠子上。(C) 磁珠洗涤和蛋白质回收。用 80% (v/v) 乙醇洗涤珠子两次,用乙腈洗涤一次,然后与 20 μL 0.05% (w/v) SDS/100 mM ABC 混合,以从珠子中回收蛋白质;搅拌 5 分钟后,用磁铁去除磁珠,并用 AX-StageTip 纯化所得蛋白质溶液。SP3磁珠请参考 http://www.purimagbead.com/Product/1736805216.html
将吸附在磁力架上的磁珠风干 10 分钟。传统的 PEPPI 工作流程使用 (1) PEPPI-MCW 与 MCW 沉淀或 (2) PEPPI-FASP 结合 FASP 和 AnExSP。PEPPI-MCW 从电泳到纯化的时间为 2.3 小时,PEPPI-FASP 为 5.3 小时,而 PEPPI-SP3 需要 3.4 小时(图 S5);FASP 是一个相对耗时的过程,通过使用 SP3 已得到极大的改进。
接下来,我们通过与 PEPPI-MCW 和 PEPPI-FASP 的比较验证了 PEPPI-SP3 的性能。根据相应的工作流程处理来自 10 μg HCPE 的 PEPPI 级分,并使用纳米 LC-MS 通过 DIA 进行 SDS-PAGE 和定量蛋白质组学。图 3 显示了从每个工作流程获得的 PEPPI 级分的 SDS-PAGE 结果,CBB 染色图像显示,在 100 kDa 以上的高 MW 区域和低于 20 kDa 的低 MW 区域,工作流程之间存在很大差异。我们以前的研究已经表明,AnExSP 不适用于高 MW 蛋白;与 MCW 相比,使用 AnExSP 进行蛋白质纯化(FASP 和 SP3)的工作流程对 100 kDa 以上的蛋白质的回收率往往较低。在低于 20 kDa 的 MW 区域,已知通常会导致低 MW 蛋白回收损失的 MCW 加工显示,与 FASP 和 SP3 相比,回收的蛋白质量预期减少。最后,比较 FASP 和 SP3,注意到用 SP3 处理的馏分具有比 FASP 更高的谱带密度,表明 SP3 在小于 20 kDa 的区域具有绝对优势,这是当前 TDP 分析的主要目标区域。通过 ContamSpot 分析评估纯化级分中 SDS 的水平,显示 SP3 处理的级分的 SDS 水平低于 0.002% (w/v),并且纯化结果与 MCW 相当(图 S6)。即使将 HCPE 从 10 μg 增加到 40 μg(图 S7),每个工作流程对馏分的纯化结果与 10 μg 时的结果相似,SP3 在 20 kDa 以下的性能明显好得多。
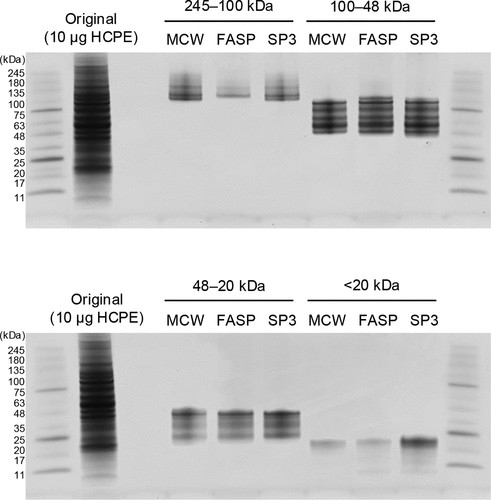
图 3.通过三种不同方法纯化的 PEPPI 组分的 SDS-PAGE 图像。通过三种不同的方法(MCW、FASP和SP3)纯化来自10 μg HCPE的四种PEPPI组分(245-100 kDa、100-48 kDa、48-20 kDa;<20 kDa),并通过SDS-PAGE再次分离这些组分。凝胶分离的组分用 CBB 染色。
对于 DIA 分析,将组分合并,用胰蛋白酶/Lys-C 消化,并进行全蛋白质组定量(图 4A)。我们的分析在所有三个工作流程中共检测到 5379 个蛋白质(表 S2),每个工作流程中检测到的数量几乎没有差异(图 4B),并且在所有三个工作流程中都检测到许多组分 (5016)。图 S8 显示了两种不同工作流程(SP3 与 MCW 或 SP3 与 FASP)之间 DIA 定量的比较结果:在 SP3 与 MCW 的情况下,每种方法显示显著变化的蛋白质数量(>3 倍,p < 0.05)相似(MCW:229 和 SP3:197)。在这些蛋白质中,在 MCW 中检测到的蛋白质显示更多氨基酸长度较长的蛋白质,而在 SP3 中检测到的蛋白质显示更多氨基酸长度较短的蛋白质(<300 个氨基酸)(图 4C),这一趋势与 SDS-PAGE 结果中观察到的趋势一致。在 SP3 与 FASP 的情况下(图 S8),显示显著差异的蛋白质在 SP3 中的丰度是 FASP 中的三倍(FASP:76 和 SP3:220),并且 SP3 中丰富的大多数蛋白质是氨基酸残基少于 300 个的蛋白质(图 4C)。这些结果表明,当目标蛋白质是低 MW 蛋白时,SP3 是首选工作流程。事实上,当考虑组蛋白(TDP 中蛋白质形式分析的重要靶标)时,SP3 对本研究中检测到的所有 15 种组蛋白都显示出比其他两种更好的恢复结果(图 S9)。相比之下,对于 TDP 分析仍未开发的高 MW 蛋白,MCW 将是首选工作流程。我们还使用 DIA 数据评估了每个工作流程的定量准确性。图 4D 显示了小提琴图中每个工作流程的变异系数 (CV) 分布,表明 SP3 在定量准确度方面优于其他工作流程。与其他工作流程相比,FASP 的定量精度较低,这可能是由于超滤过滤器上的蛋白质吸附损失所致。
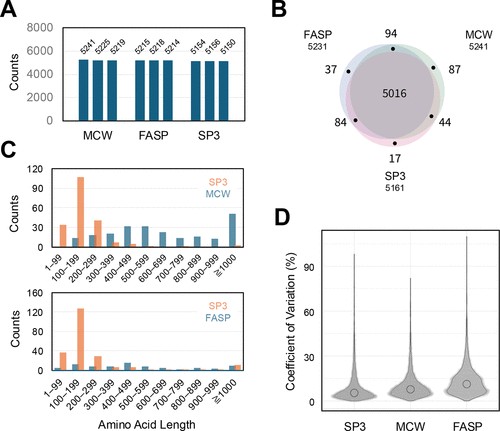
图 4.通过 DIA 定量评估不同的 PEPPI 工作流程。(A) 使用 PEPPI-MCW (MCW)、PEPPI-FASP (FASP) 和 PEPPI-SP3 (SP3) 三种 PEPPI 工作流程从 10 μg HCPE 中鉴定的蛋白质的比较,混合赖斯-C/胰蛋白酶,并通过 LC-MS 进行 DIA 分析。(B) 维恩图显示了每个工作流程中鉴定的蛋白质之间的关系。(C) 蛋白质中氨基酸长度的比较,如图 S8 所示的显著差异。(D) 每个工作流程中鉴定的蛋白质组的 CV 分布。圆圈表示中位数。
PEPPI-SP3 组分的 TDP 分析
接下来,我们通过将 PEPPI-SP3 方法与 PEPPI-MCW 和 PEPPI-FASP 方案进行比较,验证了 PEPPI-SP3 方法对 TDP 的性能。为了进行此比较,对 40 μg HCPE 的三个技术重复进行 PEPPI 分级,以生成每种处理方法的 0-30 kDa 馏分(图 S10)。在用相应的方法(MCW、FASP 和 SP3)处理后,使用 FAIMS-HiHi 方法分析样品,该方法依赖于内部补偿电压 (CV) 步进,(32) 三次运行中的每一次都使用三个不同的 CV 值(即,每个样品总共经受 9 个 CV)以增加所研究蛋白质组的深度。
总体而言,PEPPI-SP3 方法在多个指标上表现出优于其他两种方法的显著优势。相对于 MCW 方法,它显著增加了 60% 的 UniProt 种质数量和 85% 的蛋白质组鉴定数量(图 5A)。虽然与 MCW 方法相比,FASP 方法确实增加了鉴定结果,但它比 SP3 的鉴定数量少了约 20%。根据对 −40 V 补偿电压的总离子色谱图 (TIC) 的目视检查,鉴定数量的差异并不奇怪,该 CV 值导致每种方法鉴定出大多数蛋白质形式(图 S11)。通过将全局 TIC 强度归一化为 SP3 样品,FASP 样品显示信号降低 ∼ 35%。然而,当在 SP3 和 MCW 之间进行类似的比较时,尽管 MCW 样品的负载量高出 75%,但 MCW 的最大强度降低了 ∼50%。这表明与 FASP 和 SP3 方法相比,MCW 的蛋白质组回收率较低。此外,SP3 处理样品的复杂性(根据色谱图中存在的不同洗脱峰的数量估计)高于其他两种处理方法中的任何一种。
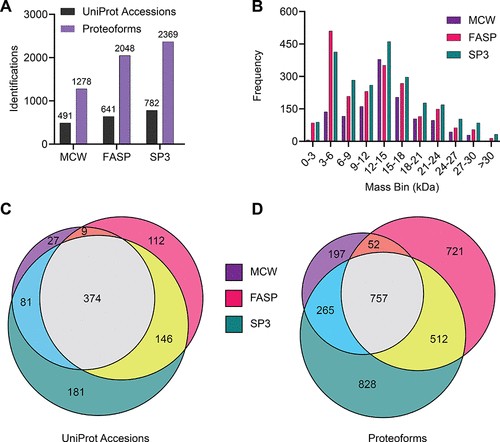
图 5.三种治疗方法的比较总结。(A) 已鉴定的 UniProt 登录号和独特蛋白形式的全局计数。(B) 已鉴定的蛋白质形式的质量分布。(C) 已识别的 UniProt 登录号的维恩图。(D) 已鉴定的蛋白质形式的维恩图。三种治疗方法的结果根据图中的图例进行颜色编码
我们对三种治疗方法之间主要差异的调查揭示了 FASP 和 SP3 方法的独特优势。一般来说,SP3 方法在整个 0–30 kDa 质量范围内优于 MCW 和 FASP 方法(图 5B)。唯一值得注意的例外是 3–6 kDa 质量 bin,其中 FASP 方案鉴定出的蛋白质形式多出 25%。FASP 和 SP3 方法对低于 9 kDa 的蛋白质形式具有显著优势,导致在该范围内鉴定的蛋白质形式比 MCW 处理的样品多 >200%。在聚合 0-9 kDa 范围内,FASP 方案鉴定的蛋白质形式略多于 SP3 方案(分别为 803 和 785)。然而,在较高分子量范围 (>15 kDa) 下,SP3 方法与 MCW 方案相比保持了相当大的优势(鉴定结果提高了约 80%),而以前在研究人血清蛋白质形式时,MCW 方法的性能与 FASP 方案相当。在相同的 15–30 kDa 质量范围内,SP3 样品中鉴定的蛋白质形式比 FASP 样品多 30%(表 S3–S5)。
虽然 PEPPI-SP3 方法在 0-30 kDa 范围内实现了更多的鉴定,但我们想确定三种方法鉴定之间的重叠程度。在查看鉴定出的 UniProt 种质数量时,SP3 方法鉴定出 MCW 方法鉴定的 93% 和 FASP 方法鉴定的 81%(图 5C)。然而,当我们进入蛋白质组水平时,我们观察到这三种方法之间具有更高程度的唯一性。在 MCW 和 FASP 样品中鉴定出的与 SP3 相同的蛋白质形式的百分比分别降低到 80% 和 62%(图 5D)。
接下来,我们检查了质谱分析的技术变异性。我们首先比较了给定处理方法的技术重复的色谱图。在检查各种 CV 时,目视检查没有显示技术重复之间的实质性差异(图 S12A 中可以找到 SP3 技术重复的 CV – 40 V 示例)。由于色谱图的差异很小,我们研究了每种处理方法的 MS 运行之间蛋白质组鉴定的重叠情况(图 S12B)。在比较每次 MS 运行中每种处理方法的所有三个技术重复中鉴定出的蛋白形式数量时,我们观察到 MCW 技术重复在其三个技术重复中共享的已鉴定蛋白质形式的百分比略高于 FASP 或 SP3 技术重复。每次 MS 运行都观察到此趋势。我们将技术变异性至少部分归因于 DDA 中前体选择的随机性,以及用 FASP 和 SP3 方法处理的样品复杂性增加(即,较大的蛋白质形态异质性,较低的鉴定重现性)。可以使用多种措施来减轻这一限制,其中包括专门增加基于分子量的 PEPPI 馏分的数量以进行分析和/或使用更长的色谱梯度。使用 SP3 方法处理的单个样品的 TIC 痕量具有高度重现性,并且与 MCW 样品相比,在所有重复中鉴定的蛋白质形式分数仅略有降低,这表明 SP3 方法可能适用于基于无标记蛋白质形式定量的定量 TDP 研究。
虽然质谱水平的技术差异可以解释高度唯一性的一些原因,但它不太可能是唯一的原因。下一步调查是确定各种治疗方法中是否存在支持或反对特定翻译后修饰的偏倚。由于三种方法之间鉴定的蛋白质形式数量存在很大差异,因此我们将给定 PTM 的频率标准化为鉴定出的蛋白质形式数量,以产生每 100 个识别蛋白质形式的比率。然后,我们根据两种方法之间比率的 Log2 转换对 10 个最常见识别的 PTM 进行了成对比较(图 S13)。综上所述,FASP 方法比 MCW 方法更能识别截断产物,但不如 SP3 方法。同时,与 MCW 和 SP3 方法相比,FASP 方法在识别蛋白质 N 端和赖氨酸的乙酰化事件以及丝氨酸、苏氨酸和酪氨酸残基的磷酸化事件方面表现出乎意料地差。即使仅考虑 C 评分为 ≥40 的蛋白质形式的 PTM 分析,也可以获得类似的结果,这些蛋白质形式是表征最好的蛋白质。这些观察结果表明,虽然 FASP 擅长识别小的蛋白质形式(即截断产物),但它难以保留对信号转导很重要的 PTM。由于 FASP 和 SP3 方法之间的差异发生在共享使用 AX-StageTip 之前,因此可以推测 FASP 中使用的离心浓缩器膜正在吸附其中一些蛋白质形式。在 PTM 鉴定方面,SP3 方法对于十种最常见的修饰,尤其是截断产物,其性能与 MCW 方法一样好或更好。通过分析所有三个数据集之间共享并携带特定 PTM 的蛋白质形式的前体离子强度,证实了这一点。结果表明,与 MCW 和 SP3 样品相比,使用 FASP 清洁的蛋白质形式的信号(特别是携带乙酰化和磷酸化)相对较弱,相反,它们显示出相当的信号强度值(图 S14)。
基于这些结果,我们试图确定其他物理化学性质是否区分了应用三种测试处理方法后鉴定的蛋白质形式。虽然通过 GRAVY 分析计算的疏水性程度在三个蛋白群之间没有显着差异,但三个组之间的等电点分布在统计学上不同 (p < 0.0001)(图 S15)。使用 MCW 方法鉴定的蛋白质形式的特点是中位数和平均 pI 分别为 9.7 和 8.9,这与使用 SP3 方法鉴定的蛋白质形式的相应值(中位数和平均 pI 分别为 9.1 和 8.4)相差不远;然而,SP3 方法也导致鉴定出更多的酸性蛋白形式 (第一个四分位数的 pI:MCW 和 SP3 分别为 7.0 和 6.4)。相比之下,FASP 方法的 pI 分布包括较少的基本蛋白质形式 (中位数和平均值分别为 7.0 和 7.3)。如图 S15B 所示,SP3 方法的 pI 分布介于其他两种方法之间,这表明这种方法能够几乎与 MCW 方法一样回收碱性蛋白质形式,同时还保留了大部分由 FASP 方法富集的中性和酸性蛋白质形式。
最后,使用 DeepTMHMM 进行的蛋白质跨膜分析表明,SP3 处理有利于恢复和鉴定预测为跨膜的蛋白质形式(占该方案获得的鉴定总数的 6.5%),优于 MCW(占总数的 5.5%),特别是 FASP(仅占总数的 2%)方法(图 6)。
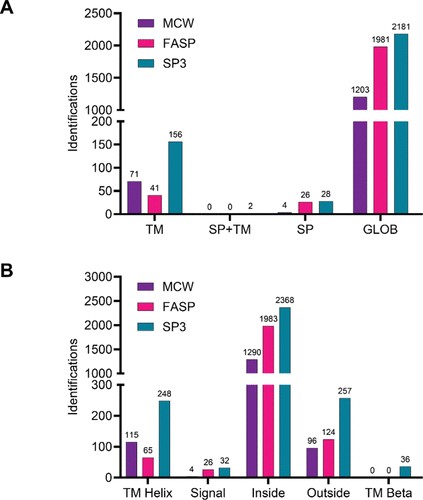
图 6.使用 DeepTMHMM 预测蛋白质形态拓扑。(A) 使用 MCW、FASP 和 SP3 去除去污剂后从样品中鉴定的蛋白质形式的拓扑类型。TM:跨膜,SP+TM:信号肽的跨膜,SP:信号肽,GLOB:球状。(B) 在 proteoforms 中鉴定的预测结构域。TM 螺旋 = α 螺旋跨膜,信号:信号肽,内部:细胞内/胞质溶质,外:ER/高尔基体/溶酶体的细胞/腔外,以及 TM Beta:β桶跨膜。一旦对 ID 总数进行标准化,FASP 处理后跨膜蛋白形式就会减少。
结论
在第一个 PEPPI 工作流程中,已经被广泛用作 TDP 的样品预分馏方法,纯化介质(通常会导致低 MW 蛋白损失)或 FASP(耗时)已被用于纯化 PEPPI 组分。在这项研究中,我们开发了 PEPPI-SP3,这是最新的和第三个 PEPPI 工作流程,它使用 SP3 微珠进行了稳定而简单的蛋白质纯化。建立回收吸附在 SP3 磁珠上的完整蛋白质的工艺对于实现 PEPPI-SP3 至关重要,这是通过将室温下快速回收与 0.05% (w/v) SDS 和使用 AX-StageTip 去除 SDS 相结合来实现的。AnExSP 将馏分成功纯化至与 MCW 相当的水平,并且通过 LC-MS 测量 TDP 时未观察到问题。PEPPI-SP3 在回收低于 20 kDa 的低分子量范围内的蛋白质方面优于传统方法,非常适合 TDP,并且由于 SP3 支持磁珠上加工,PEPPI-SP3 还可以增强 BUP 和中下蛋白质组学中的样品预处理。 尽管 SP3 在 BUP 中被确立为一种通用的样品制备方法,但它在 SDS-PAGE 后应用于凝胶中的蛋白质仍然难以捉摸。本研究中建立的 SDS-PAGE 到 SP3 通路最终允许在基于凝胶的自上而下分析中使用 SP3。
随着 PEPPI-SP3 在本研究中的成功开发,现在有三种有效的方法可用于纯化 PEPPI 组分(MCW、FASP 和 SP3),但为 TDP 选择合适的工作流程对于获得最佳结果至关重要。在作时间方面,SP3 纯化可以在比 FASP 更短的时间内完成,尽管它仍然比 MCW 慢。在可作性方面,当蛋白质量较少时,MCW 在使用中具有挑战性,因为通常很难目视确认形成的蛋白质沉淀,而且 FASP 易于执行但耗时,而 SP3 具有无论样品量如何都稳健和简单的理想组合。本研究中通过定量 DIA 分析进行的比较评估也支持 SP3 在定量准确性方面的优越性,使 SP3 成为在 TDP 中纯化 PEPPI 组分的首选工作流程,尤其是在与低 MW 蛋白(如组蛋白)的相容性方面。对于低于 30 kDa 的蛋白质形式的 TDP 分析,与 MCW 和 FASP 相比,SP3 方法提供了最多的鉴定数量,同时还能够保留与信号转导通路相关的 PTM。
尽管用当前的 TDP 很难分析超过 100 kDa 的高 MW 蛋白,但仍然可以应用 PEPPI,但对如此高 MW 的蛋白,MCW 是比 SP3 更好的选择。MCW 也可能具有成本优势,因为它不需要 SP3 微珠或超滤设备。PEPPI 的性质允许在样本数量较多时使用两种不同的方法。例如,可以在各自的 MW 范围内同时使用 SP3 和 MCW 以降低成本。但是,SP3 仍有降低成本的空间。近年来,SP4 方案通过离心而不是磁力架来回收磁珠,已被报道为 SP3 的改进方案,允许用低成本的非磁珠替换磁珠。基于低成本协议的改进将使潜在的 PEPPI-SP4 更加便宜。
TDP 的临床应用在不久的将来可能会加速,对大量样本的高通量处理的需求将相应增加。随着样品数量的增加,MCW 和 FASP 的繁琐作可能是一个主要障碍。相比之下,市售的自动化设备可用于 SP3,并且在 BUP 的多样品处理中表现出优异的性能。BUP 中 SP3 自动加工的关键技术应可转移到 TDP 的 PEPPI 分馏加工。SP3磁珠请参考 http://www.purimagbead.com/Product/1736805216.html
《使用 SP3 纯化基于凝胶的样品分级用于自上而下的蛋白质组学》总结
研究背景
1. 蛋白质组学研究目标:了解庞大的人类蛋白质组,需要开发全面的蛋白质组分析技术。
2. 自上而下蛋白质组学(TDP)的需求:自下而上蛋白质组学难以准确鉴定蛋白质形式,TDP 可直接分析完整蛋白质形式,但需样品预分馏以检测更多蛋白质组分。
3. 现有方法的问题:SDS - PAGE 是常用预分馏方法,结合 PEPPI - MS 可高效提取凝胶中蛋白质,但获得的馏分含干扰 MS 分析的 CBB 和 SDS,当前蛋白质纯化方法(如 MCW、AnExSP)存在低分子量蛋白损失、操作繁琐等问题。
实验方法
1. 材料:使用 MS 相容的人蛋白提取物,经还原、烷基化处理,调整蛋白质浓度。
2. SDS - PAGE:使用预制凝胶或自制凝胶,不同染色剂染色。
3. PEPPI 分级分离:切取染色凝胶感兴趣 MW 区域,研磨凝胶块,用含 SDS 的碳酸氢铵溶液提取蛋白质,得到 PEPPI 组分。
4. 蛋白质纯化
MCW:加入甲醇、氯仿和水,离心分层,去除上层,洗涤下层蛋白质沉淀。
FASP:通过超滤装置置换溶剂,用 AX - StageTip 纯化。
SP3:将 PEPPI 组分与 SP3 珠子和乙醇混合,吸附蛋白质,洗涤珠子,用含 SDS 的碳酸氢铵溶液回收蛋白质,再用 AX - StageTip 纯化。
5. 检测与分析
ContamSpot 检测:测定纯化后 PEPPI 组分的残留 SDS 浓度。
胰蛋白酶 / Lys - C 消化:溶解 PEPPI 组分,用胰蛋白酶 / 赖氨酸 - C 混合物消解。
NanoLC/MS/MS:获取 MS 和 MS/MS 数据,进行 DDA 和 DIA 分析。
自上而下的样品制备与分析:对 HCPE 样品进行 PEPPI 分级,用不同方法处理,通过色谱和质谱进一步分离和分析,使用 ProSight PD 等软件进行数据分析。
结果与讨论
1. 通过 SP3 纯化 PEPPI 组分
确定磁珠添加量:在 250 至 20 kDa MW 范围,磁珠添加量对回收率影响小;低于 20 kDa 时,磁珠量小于 500 μg 回收率略降,因此设定磁珠量为 500 μg。
选择蛋白质回收试剂:研究多种试剂,发现 0.05%(w/v)SDS/100 mM ABC 可在 10 分钟内从珠子中回收蛋白质,其他试剂效果不佳,最终选择 SDS 用于蛋白质回收。
2. PEPPI - SP3 工作流程
建立新流程:结合基于 SP3 的蛋白质回收和 AnExSP 纯化,建立 PEPPI - SP3 工作流程。该流程中,蛋白质组样品经 SDS - PAGE 分离、PEPPI 分级,与 SP3 珠子结合,洗涤后回收蛋白质,再经 AnExSP 纯化进行 LC - MS 分析。
流程时间比较:PEPPI - MCW 时间为 2.3 小时,PEPPI - FASP 为 5.3 小时,PEPPI - SP3 为 3.4 小时,SP3 改进了 FASP 耗时的问题。
性能验证:SDS - PAGE 结果显示,在高 MW 区域(>100 kDa),使用 AnExSP 的工作流程(FASP 和 SP3)对蛋白质回收率低于 MCW;在低 MW 区域(<20 kDa),MCW 加工导致低 MW 蛋白回收损失,SP3 处理的馏分谱带密度高于 FASP。ContamSpot 分析表明 SP3 处理的馏分 SDS 水平低于 0.002%(w/v),与 MCW 相当。DIA 分析显示,三个工作流程共检测到 5379 个蛋白质,SP3 在低 MW 蛋白回收方面表现更好,对组蛋白的恢复结果优于其他两种方法,且在定量准确度方面优于其他工作流程。
3. PEPPI - SP3 组分的 TDP 分析
鉴定数量优势:PEPPI - SP3 方法相对于 MCW 方法,显著增加了 60% 的 UniProt 种质数量和 85% 的蛋白质组鉴定数量;与 FASP 方法相比,鉴定数量多约 20%。
质量范围优势:SP3 方法在整个 0 - 30 kDa 质量范围内表现出色,在 > 15 kDa 质量范围优势明显;FASP 方法在 3 - 6 kDa 质量 bin 鉴定出的蛋白质形式多出 25%,在 0 - 9 kDa 范围鉴定的蛋白质形式略多于 SP3。
鉴定重叠情况:SP3 方法鉴定出 MCW 方法鉴定的 93% 和 FASP 方法鉴定的 81% 的 UniProt 种质;在蛋白质组水平,MCW 和 FASP 样品中鉴定出的与 SP3 相同的蛋白质形式的百分比分别为 80% 和 62%。
技术变异性:SP3 方法处理的单个样品 TIC 痕量重现性高,适用于定量 TDP 研究。
翻译后修饰(PTM)鉴定:FASP 方法在识别某些 PTM(如蛋白质 N 端和赖氨酸的乙酰化、丝氨酸等残基的磷酸化)方面表现较差,SP3 方法在 PTM 鉴定方面性能与 MCW 方法相当或更好。
蛋白质物理化学性质:SP3 方法鉴定的蛋白质形式等电点分布介于 MCW 和 FASP 之间,能回收碱性蛋白质形式,同时保留中性和酸性蛋白质形式;SP3 处理有利于恢复和鉴定预测为跨膜的蛋白质形式。
结论
1. 开发新工作流程:开发了 PEPPI - SP3 工作流程,通过室温下用 0.05%(w/v)SDS 快速回收吸附在 SP3 磁珠上的完整蛋白质,并结合 AX - StageTip 去除 SDS,实现稳定而简单的蛋白质纯化。
2. 方法优势:PEPPI - SP3 在回收低分子量(<20 kDa)蛋白质方面优于传统方法,适合 TDP,也可增强 BUP 和中下蛋白质组学中的样品预处理。
3. 工作流程选择:为 TDP 选择合适工作流程很关键,SP3 在定量准确性、操作稳健性和对低 MW 蛋白的相容性方面表现优越,是纯化 PEPPI 组分的首选工作流程;对于高 MW 蛋白(>100 kDa),MCW 是更好选择。
4. 未来展望:随着 TDP 临床应用需求增加,SP3 可借助市售自动化设备实现高通量处理,未来可通过改进降低成本,如采用 SP4 方案等。
- 上一篇:生物交联技术——交联剂功能及使用特点 2025/4/22
- 下一篇:SP3 和 SP4 捕获蛋白质的差异展现了蛋白质组学纯化技术 2025/4/15