
SP3 和 SP4 捕获蛋白质的差异展现了蛋白质组学纯化技术的机制探讨
Differences in Protein Capture by SP3 and SP4 Demonstrate Mechanistic Insights of Proteomics Cleanup Techniques,
Jessica M. Conforti, Amanda M. Ziegler, Charli S. Worth, Adhwaitha M. Nambiar, Jacob T. Bailey, Joseph H. Taube, and Elyssia S. Gallagher
Journal of Proteome Research 2024 23 (9), 3877-3889
DOI: 10.1021/acs.jproteome.4c00206
摘要:蛋白质组学实验的目标是鉴定蛋白质以观察细胞过程和疾病的变化。蛋白质组学的一个挑战是蛋白质提取后去除污染物,这可能会限制蛋白质鉴定。SP3增强样品制备是一种净化技术,其中蛋白质通过亲水相互作用液相色谱 (HILIC) 类似机制捕获在羧基修饰的颗粒上。最近的结果表明,由于蛋白质聚集机制,蛋白质被捕获在 SP3 中。SP4是一种新的净化技术,它采用蛋白质沉淀聚集来捕获蛋白质,而无需修饰颗粒。我们假设 SP3 和 SP4 捕获机制的差异会影响每种净化技术鉴定的蛋白质。在此,我们评估了使用 SP3 与 SP4 鉴定和富集的 MCF7 亚细胞组分的蛋白质,并将每种方法中的蛋白质捕获与蛋白质疏水性相关联。我们的结果表明,SP3 通过 类HILIC和蛋白质聚集机制的组合捕获更多的亲水性蛋白质,而 SP4 通过蛋白质聚集机制捕获更多的疏水性蛋白质。最终,我们证明了蛋白质捕获机制是不同的,并且选择产生高蛋白质组覆盖率的纯化技术取决于蛋白质-样品的疏水性。SP3磁珠请参考http://www.purimagbead.com/Product/1736805216.html。
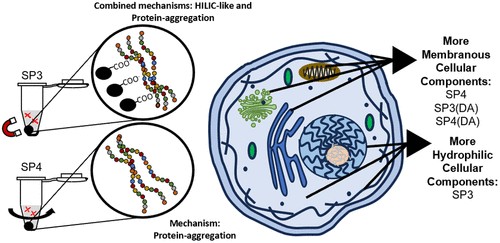
介绍
蛋白质组学实验的一个关键目标是鉴定生物样品中的蛋白质,以揭示调节细胞活性的疾病生物标志物和翻译后修饰。因此,蛋白质组学实验需要检测和鉴定具有不同特性且以不同浓度存在于生物样品中的蛋白质。通常,蛋白质组学工作流程包括蛋白质提取、二硫键还原、游离半胱氨酸烷基化、样品净化、蛋白质消化、液相色谱-串联质谱 (LC-MS/MS) 和数据库搜索。在蛋白质提取过程中,缓冲液、盐和离液剂等污染物会破坏细胞膜以释放和溶解蛋白质,同时保持生理 pH 值以避免蛋白质沉淀。虽然这些污染物对于蛋白质提取是必需的,但在电喷雾过程中会抑制消化酶活性、LC 分离度和肽电离,从而减少鉴定出的蛋白质数量。
尽管在蛋白质消解之前必须有效去除蛋白质提取产生的污染物,但样品净化仍然是蛋白质组学工作流程中的一个限制。SP3是一种净化技术,与基于过滤器的方法相比,能够鉴定更多的肽和蛋白质,具有更高的试验间重现性。SP3 使用有机变性溶剂诱导蛋白质与羧基磁珠结合。引入该技术后,有人提出蛋白质基于类亲水相互作用液相色谱 (HILIC)机制与颗粒相互作用。使用外部磁铁,将蛋白质结合的颗粒吸引到微量离心管的侧面,从而在蛋白质消化之前去除上清液中的污染物。然而,羧基磁珠可以 (1) 不充分结合蛋白质,导致残留在上清液中的蛋白质丢失;(2) 通过与蛋白酶抑制剂结合来破坏消化效率;(3) 不可逆地结合蛋白质,防止它们被带入 LC-MS/MS; (4) 如果携带到仪器中,会堵塞 LC 色谱柱。
SP3 的蛋白质捕获机制最近受到挑战,表明 SP3 主要通过蛋白质聚集机制捕获蛋白质,而不是 HILIC 样机制。SP4采用提出的这种蛋白质聚集机制,省略了磁性羧酸盐修饰的颗粒。在 SP4 中,使用有机溶剂沉淀和聚集蛋白质,使蛋白质能够通过离心沉淀。有问题的污染物留在上清液中去除,产生用于消化的聚集蛋白沉淀。与 SP3 相比,SP4 以前能够鉴定 HEK293、Jurkat 人永生化 T 和 E14 小鼠胚胎干细胞裂解物的相似或更多数量的肽和蛋白质。对于 HEK293 裂解物,与 SP3 相比,SP4 还富集了低溶解度跨膜蛋白的回收率。 蛋白质捕获的这种差异可能是由于 SP3 中使用的羧酸盐修饰颗粒。因此,我们假设 SP3 和 SP4 具有不同的蛋白质捕获机制,影响使用每种样品纯化技术时鉴定和富集的蛋白质。
在这项工作中,我们旨在比较 SP3 和 SP4 鉴定和富集的蛋白质类型。此外,我们评估了脱氧胆酸钠 (SDC) 辅助消化的能力,以对抗 SP4 产生的蛋白质沉淀的不溶性,并增加 SP3 和 SP4 中疏水蛋白质的检测。在本文中,我们比较了用 SP3、SP4、SP3(去污剂辅助 (DA))和 SP4(去污剂辅助 (DA))制备的样品的鉴定蛋白质。具体来说,我们使用密歇根癌症基金会 7 (MCF7) 乳腺癌亚细胞组分(包括细胞质、膜、可溶核、染色质结合核和细胞骨架组分)进行深度蛋白质组分析。亚细胞组分包含具有不同特性的蛋白质,并用作分级分离的一种形式,降低了全细胞裂解物的复杂性,并能够检测低丰度蛋白质。最终,我们的结果显示每种清理技术鉴定和富集的蛋白质存在差异,我们与蛋白质捕获机制的差异相关联。
实验程序
材料
碳酸氢铵来自 Sigma-Aldrich(密苏里州圣路易斯)。Emplura 无水乙醇和高纯度脱氧胆酸钠 (99%) 来自 VWR International (Radnor, PA)。胰蛋白酶/Lys-C 混合物(质谱级)来自 Promega(威斯康星州麦迪逊)。Glu-1-纤维蛋白肽 B 来自 Waters(马萨诸塞州米尔福德)。所有其他物品均购自 ThermoFisher Scientific(马萨诸塞州沃尔瑟姆)。纳米纯水来自 Purelab Flex 3 纯水系统(Elga,Veolia Environment S.A.,法国巴黎)。
细胞培养和裂解
MCF7 乳腺癌细胞(ATCC HTB-22,弗吉尼亚州马纳萨斯)在补充有 10% 胎牛血清(Gibco,Billings,MT)和 1% 青霉素/链霉素 (Gibco) 的 Dulbecco 改良 Eagle 培养基(Corning,Corning,NY)中培养。使用 PlasmoTest TM 支原体检测试剂盒(InvivoGen,San Diego,CA)定期检测细胞是否受到支原体污染,并在 80% 汇合时传代。
使用含有 0.025 M Tris、0.15 M NaCl、0.001 M EDTA、1% NP-40 和 5% 甘油的裂解缓冲液(Pierce 免疫共沉淀试剂盒,ThermoFisher Scientific)收获全细胞蛋白。使用培养细胞亚细胞蛋白分级分离试剂盒 (ThermoFisher Scientific) 对 1.0 × 107 个细胞进行亚细胞蛋白分级分离。使用二辛可宁酸 (BCA) 测定法测定蛋白质浓度。通过使用每个亚细胞组分的已建立蛋白质标志物进行免疫印迹来确认亚细胞组分分离。
还原和烷基化
将蛋白质样品加入低结合力的 1.5 mL 微量离心管中,并在 Reacti-Therm 单模块加热模块 (ThermoFisher Scientific) 上在 100 °C 下使蛋白质变性 5 分钟。在室温下,将二硫苏糖醇 (DTT) 添加到蛋白质样品中(蛋白质与 DTT 摩尔比为 1:10),并将样品在 60 °C 和 1,000 rpm 下在 Eppendorf ThermoMixer 温度控制装置(Eppendorf,Hamburg,Germany)上孵育 30 分钟。然后加入碘乙酰胺 (IAA)(蛋白质与 IAA 摩尔比为 1:100),并将样品在室温下避光孵育 30 分钟,以使游离半挂烃烷基化。用额外的 DTT(5:1 DTT 与 IAA 摩尔比)淬灭烷基化反应。
SP3增强样品制备
SP3磁珠请参考http://www.purimagbead.com/Product/1736805216.html。Speedbead磁性羧酸盐改性颗粒(GE45152105050250 和 GE65152105050250,GE Healthcare,芝加哥,伊利诺伊州)以 1:1 (v/v) 的比例合并,用水洗涤 3 次,并以终浓度 50 mg/mL 重悬于水中。将制备的颗粒以 10:1 (w/w) 的颗粒与蛋白质比例添加到蛋白质样品中,并以 4:1 (v/v) 的乙腈与蛋白质比例加入冷却的乙腈 (4 °C) 以诱导与颗粒结合。将蛋白质样品在 ThermoMixer 上于 24 °C 和 1000 rpm 孵育 5 分钟以增强颗粒-蛋白质结合,然后在室温下置于 1 T 定制磁架上 5 分钟。将带有结合蛋白的磁性颗粒拉到微量离心管的一侧,并将上清液中的污染物作为废液去除。用 80% 乙醇水溶液 (v/v) 洗涤蛋白质-颗粒混合物,置于磁力架上 5 min,含污染物的上清液作为废液除去。
SP4增强样品制备
以 4:1 (v/v) 的乙腈与蛋白质比例加入冷冻乙腈 (4 °C),以溶解和聚集蛋白质。将样品涡旋混合 (<500 rpm) 5 秒,然后以 16,000g 离心 10 分钟,以使用 Fisherbrand accuSpin Micro 17 微量离心机 (ThermoFisher Scientific) 沉淀蛋白质。除去含污染物的上清液作为废液,用 80% 乙醇水溶液 (v/v) 洗涤蛋白沉淀,以 16,000g 离心 10 min,含异物的上清液作为废液除去。
标准消解
胰蛋白酶/赖氨酸-C 以 1:50 (w/w) 的酶蛋白比例加入 100 mM 碳酸氢铵,pH 值为 8。将蛋白质在 ThermoMixer 上于 37 °C 和 1000 rpm 消化 18 小时。然后将肽样品置于磁力架 (SP3) 上或以 16,000g (SP4) 离心 10 分钟,并将含肽的上清液移至干净的样品瓶中。
SP3 (DA) 和 SP4 (DA)去污剂辅助消化
胰蛋白酶/赖氨酸-C 以 1:50 (w/w) 的酶蛋白比例加入 100 mM 碳酸氢铵 (pH 8) 和 1% SDC(使用 10% SDC (w/v) 储备液)。蛋白质在 ThermoMixer 上以 37 °C 和 1,000 rpm 消化 18 小时。将乙腈加入 20% (v/v) 的最终溶剂混合物中以提高疏水肽回收率,并将三氟乙酸 (TFA) 加入至 0.5% 以沉淀 SDC 并淬灭消化。将肽样品以 16,000g 离心 10 分钟,以沉淀去污剂和任何不溶性碎片。对于 SP3 (DA),然后将样品放在磁力架上 5 分钟,以确保颗粒不会被带入仪器。然后将 SP3 (DA) 和 SP4 (DA) 样品的肽上清液移至干净的样品瓶中。总结了所有四种样品清理技术。SP3磁珠请参考http://www.purimagbead.com/Product/1736805216.html。
Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS)
根据 Thermo Scientific Nanodrop One 的测量结果,将肽用 99% 水、0.9% 乙腈和 0.1% 甲酸稀释至浓度约为 ∼0.5 μg/μL。将肽(2 μL)进样到配备Waters ACQUITY UPLC M-Class Symmetry C18捕集柱(5 μm, 180 μm × 20 mm)的Waters nanoACQUITY UPLC色谱柱上,与Waters ACQUITY UPLC PST BEH C18 nanoACQUITY分析柱(1.7 μm、75 μm×100 mm)联用。在 MS 分析之前,使用由溶剂 A(99.9% 水和 0.1% 甲酸)和溶剂 B(99.9% 乙腈和 0.1% 甲酸)组成的梯度来分离肽。完整的 LC 梯度如表 S2 所示。
使用Fossilion Sharp Singularity LOTUS nESI发射器(Fossiliontech,西班牙马德里),通过Waters Z-Spray NanoLockspray电离源,将从液相色谱柱洗脱的肽喷入Waters Synapt G2-S高分辨率质谱仪中。使用 Glu-1-纤维蛋白肽 B 作为参考锁定质量,精确 m/z 为 785.8421,用于质量校准和采集后质量校正。在正离子分辨率模式下,以下串联 MS 参数用于数据非依赖性分析:质量分辨率:20,000,扫描范围:50–2000 m/z,超清晰 MSE (UDMSE) 的传递碰撞能量斜坡:歌涡分箱 0–20,保持 17 eV;移动性箱 20-110,将碰撞能量从 17 eV 增加到 45 eV;移动性 bin 110–200,将碰撞能量从 45 eV 增加到 60 eV。优化的 MS 参数如图 S6 和表 S3 所示。
数据库搜索
用于蛋白质组学的 Progenesis QI(4.2 版,非线性动力学,沃特世公司)用于搜索含有 80,027 种蛋白质(经典蛋白和亚型,2022 年 6 月下载)的 Uniprot Homo Sapien 参考蛋白质组 (UP000005640) 的实验数据。为了确定仅用一种方法鉴定的肽和蛋白质的数量,我们检索了来自单一纯化技术的数据。而成对比较 (SP3 与 SP4、SP3 与 SP3 (DA) 和 SP4 与 SP4 (DA)) 通过每种方法进行火山图、水肿总平均数 (GRAVY) 和基因本体论 (GO)-Slim 分析,以确定富集的蛋白质。导入数据,校正 Glu-1-纤维蛋白肽 B 锁质量数,并启用离子计数工作流程,以使用 UDMSE 鉴定肽。使用以下搜索参数:肽和片段容差:自动,酶特异性:赖氨酸和精氨酸 C 端裂解 (/K-\P/R-\P),最大离子电荷:20,漏缺切割:最多 2,运行对齐:自动,峰挑选灵敏度:自动,最大蛋白质质量:250 kDa,离子匹配要求:5 个片段/蛋白质、2 个片段/肽和 1 个肽/蛋白质。错误发现率设定为 <1%,iadbs.exe Progenesis Qi 用于蛋白质组学,“动态”创建随机序列。修饰包括半胱氨酸的固定脲甲基化、蛋氨酸的可变氧化以及丝氨酸、苏氨酸和酪氨酸的可变磷酸化。磷酸化被包括在内,因为这种常见的修饰对细胞功能至关重要,并且在乳腺癌发病机制中特别发现。使用 Hi-3 和蛋白质分组完成相对定量的自动处理,以生成原始和归一化的蛋白质丰度、最大倍数变化和方差分析值。所有 LC-MS/MS 原始数据均已存入 MassIVE 和 ProteomeXchange 存储库,并带有以下入藏号:(MSV000094130) 和 (PXD049965)。
统计学
对于每个生物样品(全细胞裂解物、细胞质、膜、可溶核、染色质结合核和细胞骨架组分),为每种净化技术(SP3、SP4、SP3(DA) 和 SP4(DA))制备三个样品制备重复,共 72 个样品。每个样品完成 3 次技术仪器重复,共计 216 次仪器运行。除非另有说明,否则所有表格、图表和条形图都使用 Microsoft Excel(版本 2301)。成对比较的统计显着性由双尾 F 检验和 t 检验确定。在 Excel 中按描述手动搜索鉴定出的蛋白质组,以确定它们是否在一种或多种样品纯化技术中被鉴定出来。UpSet 图用于可视化蛋白质组交叉,并使用 UpSetR 包在 R 中构建。DAVID 功能注释工具 (2021 版,2023 年 2 月访问)用于将蛋白质组 中的先导蛋白鉴定映射到细胞成分。将 DAVID 输出的细胞成分手动分为以下几类:细胞质、膜、细胞核、细胞骨架或非特异性成分。非特异性细胞组分包括无法有效归类为其他类别的一般注释,包括“细胞”、“细胞部分”、“细胞内细胞器”等。图 S7 和 S8 描述了 DAVID 输入参数以及 DAVID 的所有细胞组分输出,以及它们如何被分组到类别中。然后构建饼图,显示映射到每个细胞类别的已鉴定蛋白质的百分比。在 Excel 中使用 Progenesis 的最大倍数变化和 P 值 (ANOVA) 输出构建火山图,以观察每种样品净化技术富集的蛋白质组。使用 GRAVY 计算器 (https://www.gravy-calculator.de/index.php) 完成 GRAVY 分析,对火山图中富集的蛋白质组进行铅蛋白鉴定 。PANTHER (版本 17.0,2023 年 2 月访问)用于对 Progenesis 中富集的蛋白质组进行 GO-Slim 细胞成分分析。所有 PANTHER 输入参数如图 S9 所示。
结果与讨论
根据亚细胞组分的不同,在不同的样品纯化技术中观察到肽和蛋白质组鉴定的增加
我们首先比较了 MCF7 细胞裂解物的每种纯化方法中鉴定出的肽和蛋白质组的数量。与以前的研究一致,在比较所有四种纯化技术时,SP4 能够鉴定出全细胞裂解物中数量最多的肽和蛋白质组(图 1)。与过去的参考文献相比, 全细胞裂解物样品的蛋白质鉴定似乎较少。这可能是由于用于测定样品浓度的 Nanodrop 测量存在不确定性,因此向 LC-MS/MS 中进样的样品量较少。然而,我们预计影响蛋白质鉴定的最重要因素是在工作中使用飞行时间 (TOF) 质量分析仪。众所周知,全细胞裂解物样品的复杂性会导致 TOF 质量分析器的鉴定计数降低,即使优化了离子淌度(图 S6)和 Tune 页面参数(表 S3),这是由于低丰度蛋白质的掩盖、光谱复杂性增加和肽电离效率的差异。本研究中使用了 TOF 质量分析器,因为它能够通过数据非依赖性采集来采集数据,并通过数据依赖性采集分析样品中的所有离子与选定的离子。为了增加蛋白质组覆盖率以对 MCF7 乳腺癌细胞中的低丰度蛋白质进行深度分析,使用了亚细胞组分离。总体而言,SP3、SP4、SP3(DA) 和 SP4(DA) 在所有亚细胞组分中分别鉴定出 3451、3517、3070 和 3094 个独特蛋白组。与之前的研究一致,当从组合的亚细胞组分中汇集鉴定出的蛋白质组时,我们看到鉴定结果增加。
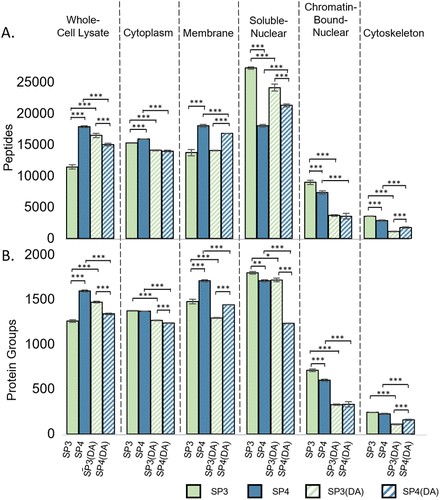
图 1.使用 SP3、SP4、SP3(去污剂辅助 (DA))和 SP4(去污剂辅助 (DA))对 MCF7 乳腺癌全细胞裂解物和亚细胞组分的鉴定肽 (A) 和蛋白组 (B) 的平均数量进行比较。样本由虚线分隔,图顶部带有标签。误差线表示均值的标准误差。条形图上方的括号线显示成对比较的显著性,用 *p < 0.05、**p < 0.01 和 ***p < 0.001 表示。没有 * 表示法的比较没有显著差异,并且处于测量不确定度范围内。
预计亚细胞组分具有较高的亲水性或疏水性蛋白质的普遍性;因此,亚细胞组分用于评估蛋白质捕获机制的差异。首先评估膜组分的鉴定肽和蛋白质组的数量。与所有方法相比,SP4 鉴定出膜组分中数量最多的肽和蛋白质组,其中包括包含疏水结构域的膜蛋白(图 1)。
然后比较已鉴定的肽和蛋白质组的数量,以查找预期含有亲水性蛋白质的组分,包括细胞质、可溶性核、染色质结合核和细胞骨架组分。对于细胞质组分,SP3 和 SP4 产生最多的已鉴定蛋白组,而 SP4 产生最多的肽(图 1)。对于可溶性核和染色质结合的核组分,SP3 提供了最多的肽和蛋白质组鉴定。最后,对于细胞骨架部分,SP3 产生最多的鉴定肽,而 SP3 和 SP4 都鉴定出大量蛋白质组。当考虑到亲水性蛋白(如肌动蛋白丝和微管)和疏水蛋白(包括中间丝,如角蛋白、波形蛋白和神经丝)都存在于该组分中时,预计 SP3 和 SP4 鉴定的蛋白质组的数量相似。以前,与 SP4 相比,SP3 在细胞裂解物中产生的鉴定肽和蛋白质数量较少。然而,此处介绍的结果表明,SP3 是一种有效的净化技术,用于识别某些亚细胞组分中相似或更高数量的蛋白质组,特别是那些预计含有高丰度亲水性蛋白质的组分。
在评估标准品与去污剂辅助消化时,除了使用 SP3 与 SP3 (DA) 的全细胞裂解物进行比较外,SDC 辅助消化没有产生最高数量的鉴定肽和蛋白质组鉴定(图 1)。预计 SDC 不会对肽和蛋白质组鉴定产生负面影响,因为它是在 LC-MS/MS 之前沉淀的(图 S3)。然而,先前的报道表明,一些肽在消化后的酸化过程中会与 SDC 沉淀,从而减少检测到的肽的总数。
不同的样品净化技术可识别不同类型的蛋白质
接下来,我们比较了使用注释细胞成分的每种清理技术鉴定的蛋白质类型。根据它们是使用一种、两种、三种还是所有四种纯化技术鉴定,对所有鉴定的蛋白质组进行排序(图 2)。评估通过所有纯化技术鉴定的蛋白质,通过所有四种方法分别鉴定出全细胞裂解物、细胞质、膜、可溶核、染色质结合核和细胞骨架组分的 484、515、572、330、199 和 50 种蛋白质(图 2)。然后通过 DAVID 功能注释工具将所有纯化技术鉴定的蛋白质组的先导蛋白鉴定映射到细胞成分类别。当评估通过所有四种方法在每个组分中鉴定的蛋白质时,观察到与每个细胞成分相关的鉴定蛋白质增加,表明亚细胞分级分离是成功的(图 S10)。然而,对于所有样品和亚细胞组分,使用所有四种纯化技术鉴定出不到 20% 的检测到的蛋白质组,这表明基于纯化技术鉴定的蛋白质组存在明显差异。
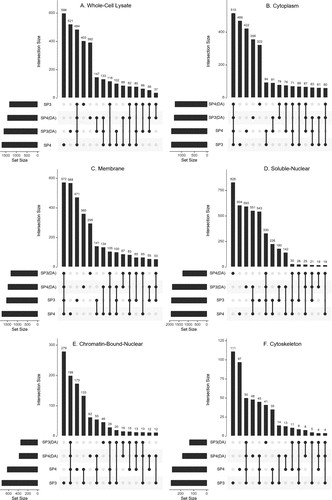
图 2.使用 SP3、SP4、SP3(DA) 和 SP4(DA) 对全细胞裂解物 (A)、细胞质 (B)、膜 (C)、可溶性核 (D)、染色质结合核 (E) 和细胞骨架 (F) 组分的已鉴定蛋白组数的异常图。组大小表示每种方法的已鉴定蛋白质组的总数。条形图显示了用 1 种、2 种、3 种或全部4种方法鉴定蛋白质组的频率,如条形图下方的黑色实线圆圈所示。
然后,我们专注于比较仅使用一种纯化技术鉴定的蛋白质。首先评估全细胞裂解物和膜组分,预计它们含有疏水蛋白。我们的结果表明,与这两个样品的其他纯化技术相比,仅通过 SP4 鉴定的蛋白质数量更多(图 2)。与所有纯化技术相比,SP4 (DA) 产生的蛋白质在全细胞裂解物中映射到最高百分比的膜细胞成分 (40%) (图 3Aiv),而 SP4 产生的蛋白质在膜部分中映射到最高百分比的膜细胞成分 (41.5%) (图 3Cii)。这些结果表明,对于预期含有疏水性蛋白质的样品,与 SP3 和 SP3(DA) 相比,仅通过 SP4 和 SP4(DA) 鉴定的膜蛋白更多。
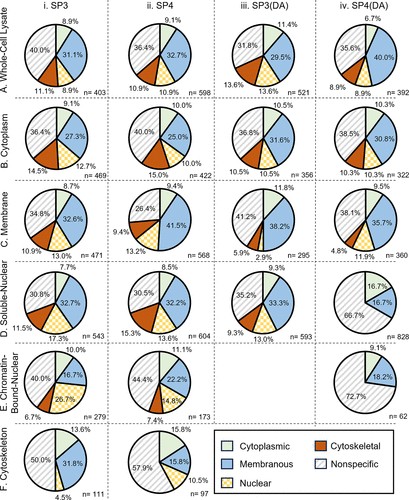
图 3.每个样品仅通过一种纯化技术鉴定的蛋白质组的细胞组分类别(列:SP3 (i)、SP4 (ii)、SP3(DA) (iii)、SP4(DA) (iv))(行:全细胞裂解物 (A)、细胞质 (B)、膜 (C)、可溶性核 (D)、染色质结合核 (E) 和细胞骨架 (F) 级分)。通过 DAVID 功能注释工具将仅由一种纯化技术鉴定的蛋白质组 (n) 映射到细胞组分,并手动分组为细胞组分类别:细胞质(纯绿色)、膜(纯蓝色)、细胞核(方格黄色)、细胞骨架(纯红色)或非特异性(条纹灰色)。缺失饼图表示使用指定方法在该亚细胞组分中唯一鉴定的蛋白质没有显著的功能注释。类别百分比的计算方法是将类别中映射的细胞成分的数量除以映射细胞成分的总数。
对于含有亲水性蛋白质(细胞质、可溶性核、染色质结合核和细胞骨架组分)的组分,还评估了仅用一种纯化技术鉴定的蛋白质。在细胞质、染色质结合的核和细胞骨架组分中,与其他纯化技术相比,仅由 SP3 鉴定的蛋白质数量更多(图 2)。对于细胞质组分,SP3 (DA) 产生最高百分比的映射到细胞质细胞成分的蛋白质 (10.5%)(图 3Biii)。对于可溶性核和染色质结合的核级分,SP3 产生的映射到核细胞成分的蛋白质百分比最高(分别为 17.3% 和 26.7%)(图 3Di,Ei)。这些结果表明,对于含有亲水性蛋白的样品,与 SP4 和 SP4(DA) 相比,仅用 SP3 和 SP3(DA) 鉴定的细胞质和细胞核蛋白更多。
成对比较可确认富集蛋白的差异
到目前为止,我们已经比较了使用每种纯化技术检测到的蛋白质组的数量和身份的差异。然而,比较通过两种方法捕获但观察到具有显著丰度差异的蛋白质组也很重要,我们将其称为富集差异。为了评估富集的蛋白质组,使用成对比较 (SP3 与 SP4、SP3 与 SP3 (DA) 和 SP4 与 SP4 (DA)) 生成火山图。
首先比较 SP3 与 SP4 样品纯化技术,以确定每种方法富集的蛋白质组数。在评估含有疏水性蛋白的全细胞裂解物和膜组分时,与 SP3 相比,SP4 从全细胞裂解物(图 4Ai)和膜组分(图 4Ci)中富集了更多的蛋白质组。虽然富集的蛋白质数量仅占总鉴定结果的一小部分(图 1),但 SP3 或 SP4 富集的蛋白质百分比与之前对全细胞裂解物的研究一致。此外,这些结果与以前的研究一致,这些研究表明,与 SP3 相比,SP4 在全细胞裂解物中富集了更多的蛋白质。
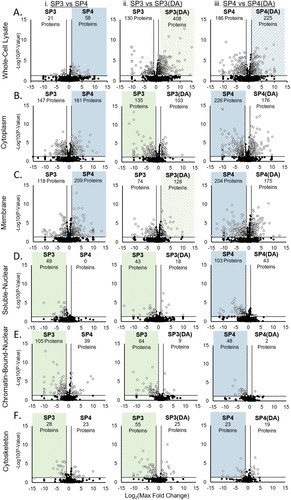
图 4.Volcano 图比较了全细胞裂解物 (A)、细胞质 (B)、膜 (C)、可溶性核 (D)、染色质结合核 (E) 和细胞骨架 (F) 组中 SP3 与 SP4 (DA) 之间富集蛋白组的数量 SP4 与 SP4 (DA) (ii) 以及 SP4 与 SP4 (DA) 之间的富集蛋白组数。比较中每种方法的富集蛋白质组数(最大倍数变化> 2,P 值< 0.05)显示在每个面板的顶部。突出显示的面板表示在每次成对比较中产生最高富集蛋白组数的方法。
比较 SP3 与 SP4 对于预期含有亲水性蛋白质的亚细胞级分,SP3 在染色质结合的核(图 4Ei)和细胞骨架级分(图 4Fi)中富集了更多的蛋白质组。与由 SP4 富集的 0 个蛋白组相比,SP3 还富集了更多的可溶性核组分蛋白组(图 4Di)。对于细胞质部分,与 SP3 相比,SP4 富集了更多的蛋白质组(图 4Bi)。然而,有许多蛋白质组同时被 SP3(147 个蛋白质组)和 SP4(181 个蛋白质组)富集(图 4Bi),表明 SP3 和 SP4 富集了细胞质组分中的不同蛋白质。因此,与 SP4 相比,SP3 丰富了细胞核和细胞骨架组分中更多蛋白质基团的鉴定。
还比较了标准酶解与去污剂辅助消化的富集蛋白组数。虽然去污剂辅助消化不会产生最多的肽和蛋白质组鉴定(图 1),但与 SP3 相比,SP3(DA) 在全细胞裂解物(图 4Aii)和膜(图 4Cii)组分中富集了更多的蛋白质组,而与全细胞裂解物中的 SP4 相比,SP4(DA) 富集了更多的蛋白质组(图 4Aiii)。此外,在预期含有更多亲水性蛋白质的亚细胞级分中,与去污剂辅助消化相比,标准消化富集了更多的蛋白质组(图 4Bii、Dii、Eii、Fii、Biii、Diii、Eiii、Fiii)。这些结果表明,对于含有亲水性蛋白质的馏分,与去污剂辅助酶解相比,标准酶解富集了更多的蛋白质组。
通过每种纯化技术富集的蛋白质都依赖于疏水性
到目前为止,我们的数据表明,每种纯化技术捕获的蛋白质都取决于蛋白质疏水性。为了确认 SP3 富集亲水性更强的蛋白质,而 SP4 富集更疏水性的蛋白质,计算了 GRAVY 评分,用于从火山图中富集蛋白组的先导蛋白鉴定(图 4)。将富集蛋白的 GRAVY 评分绘制为频率分布,以比较不同净化方法之间疏水性的差异。亲水性或疏水性更强的蛋白质的 GRAVY 评分分别转移到图中更负或更正的区域。大多数蛋白质包含亲水性和疏水性结构域,因此预计许多 GRAVY 评分在 -0.2 到 0.2 之间。在比较 GRAVY 图时,这可能导致峰顶点的相似性; 因此,我们专注于比较 GRAVY 曲线中得分超出此范围的肩部,以检查每种方法富集的蛋白质疏水性的差异。
首先比较了 SP3 与 SP4 在全细胞裂解物和预期含有更多疏水蛋白的膜组分中的 GRAVY 曲线。对于全细胞裂解物,SP4 的 GRAVY 曲线的峰顶点为 -0.4,SP3 曲线的峰顶点为 -0.5(图 5Ai)。与 SP4 相比,SP3 在图的更负区域也有一个肩部,与 SP3 相比,在曲线的正区域由 SP4 富集的蛋白质数量更多(图 5Ai)。这些结果与之前的研究一致,表明与 SP3 相比,SP4 富含的蛋白质更具疏水性。在膜部分,SP3 和 SP4 具有相似的峰顶点,但与 SP4 相比,SP3 曲线在 GRAVY 图的更负区域有一个肩部(图 5Ci)。因此,对于全细胞裂解物和膜组分,SP3 富集了与 SP4 相似或更亲水性的蛋白质。
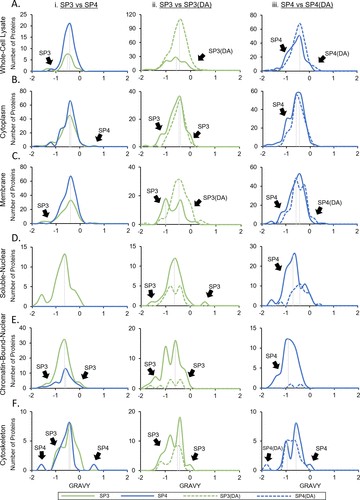
图 5.SP3 与 SP4 (i)、SP3 与 SP3(DA) (ii) 和 SP4 与 SP4(DA) (iii) 的全细胞裂解物 (A)、细胞质 (B)、膜 (C)、可溶核 (D)、染色质结合核 (E) 和细胞骨架 (F) 级分中富集蛋白组的铅蛋白鉴定的总体平均数 (GRAVY) 评分分布。负 GRAVY 值越多表示亲水性蛋白质越多,而正值越多表示疏水性蛋白质越多。垂直线表示峰顶点,以帮助比较分布。
然后比较 SP3 与 SP4 纯化技术对预期含有亲水性蛋白质的组分。对于细胞质部分,SP3 峰顶点为 -0.5,SP4 峰顶点为 -0.4。与 SP4 相比,SP3 的 GRAVY 曲线移动到图中更负的区域,并且 SP4 曲线在图的正区域有一个肩部,GRAVY 评分为 0.5(图 5Bi)。对于可溶性核级分,火山图显示与 SP3 相比,SP4 没有富集任何蛋白质(图 4Di),因此没有在 SP3 和 SP4 之间进行 GRAVY 比较(图 5Di)。对于染色质结合的核级分,SP3 峰顶点移至更负的区域,并且图的负区和阳性区都有肩部(图 5Ei)。然而,阳性区域的肩部的 GRAVY 评分在 -0.2 到 0.2 之间,表明 SP3 富集了包含亲水性和疏水性结构域的蛋白质。对于细胞骨架部分,SP3 GRAVY 曲线比 SP4 曲线更多地延伸到阳性区域,但 SP4 在非常负区捕获一个蛋白质,在非常阳性区域捕获一个蛋白质(图 5Fi)。对于该部分,与其他细胞组分相比,富集的蛋白质数量较少,并且数据显示 SP3 和 SP4 富集的蛋白质具有亲水性和疏水性蛋白质的属性。我们对预期含有亲水性蛋白质的细胞组分的结果表明,与 SP4 富集的蛋白质相比,SP3 富集的蛋白质相似或更亲水性。
富集的蛋白质是总蛋白质组鉴定的一小部分(图 1)。我们的结果表明,SP3 富集的蛋白质类似于或比 SP4 富集的蛋白质更亲水性(图 5)。这与通过单个样品净化技术捕获的蛋白质差异一致(图 3),其中我们观察到与 SP4 相比,SP3 对亲水细胞成分的蛋白质注释增加。相比之下,SP3 仅在可溶性核和染色质结合的核组分中鉴定出更亲水性的核蛋白(图 3Di,Ei),而全细胞裂解物和膜性组分中更疏水性的膜蛋白仅通过 SP4 鉴定(图 3Ai,Ci)。因此,富集蛋白质的疏水性差异(图 5)与 SP3 与 SP4 实现的蛋白质组鉴定的预期疏水性差异一致(图 3)。
还检查了比较标准和洗涤剂辅助消化的 GRAVY 曲线(图 5)。对于含有亲水性蛋白质的组分(例如细胞质、可溶性核、染色质结合核和细胞骨架组分),标准消化的 GRAVY 曲线在图的亲水区域中具有肩部。而对于含有疏水蛋白的全细胞裂解物和膜组分,在去污剂辅助消化中观察到延伸到图的疏水区域的肩部。因此,与去污剂辅助消化相比,标准消化增加了对相似或更亲水性蛋白质的检测,而与标准消化相比,去污剂辅助消化增加了对更多疏水性蛋白质的检测。
以前,我们评估了仅通过一种清理技术鉴定的蛋白质的细胞成分注释的差异(图 3)。细胞成分本体论的分析将富集的蛋白质(图 4)与执行分子功能的基因及其在细胞中的相关定位联系起来。这可以进一步了解在成对比较中差异富集的蛋白质组的生物学意义。GO 分析通过计算在样品中观察到蛋白质的概率相对于在整个人类蛋白质组中观察到蛋白质的概率,将蛋白质组映射到细胞成分本体。
首先通过 PANTHER 将来自全细胞裂解物和膜组分的火山图(图 4)的富集蛋白组映射到细胞成分本体,以比较 SP3 与 SP4。亚细胞级分的其他 GO 分析可在图 S9 中找到。在全细胞裂解物中,SP3 富集的蛋白质组定位于细胞质、膜、核和细胞骨架细胞成分本体,而 SP4 富集的蛋白质组定位于膜细胞成分本体(图 6)。预计全细胞裂解物包含所有细胞成分本体;因此,SP4 仅富集映射到膜细胞成分本体的蛋白质组这一事实进一步证实了 SP4 识别(图 1 和 3)和富集(图 4 和 5)疏水性膜蛋白的能力。同样,在膜组分中,SP3 富集的蛋白质组映射到膜和细胞骨架细胞成分本体,而 SP4 富集的蛋白质组仅映射到膜细胞成分本体(图 6)。这些结果与 SP3 鉴定(图 3Di,Ei)和富集(图 5)的亲水性蛋白的增加一致;以及 SP4 在全细胞裂解物和膜组分中鉴定(图 3Ai,Ci)和富集(图 5Ai)的疏水性膜蛋白。这些结果表明,SP3 富含具有亲水性和膜蛋白的映射到细胞成分本体的蛋白质,而仅观察到 SP4 具有疏水性蛋白富集映射到膜细胞成分本体的蛋白质。
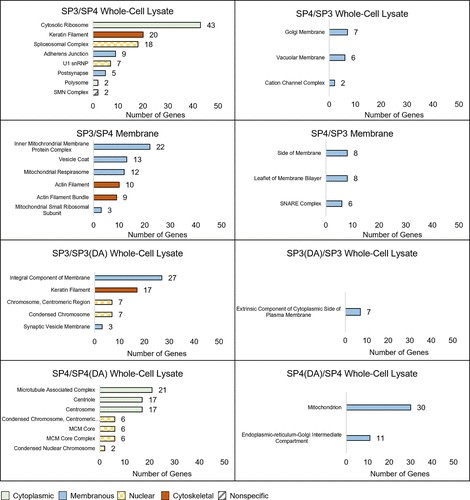
图 6.SP3 与 SP4 以及标准与去污剂辅助消化的 GO-Slim 细胞成分本体的比较。条形图表示一种清理方法与另一种清理方法中与显著富集的细胞组分本体相关的基因数量。条形颜色表示细胞成分本体的分组类别:细胞质(纯绿色)、膜(纯蓝色)、细胞核(方格黄色)、细胞骨架(纯红色)或非特异性(条纹灰色)。
还使用 GO-Slim 评估了由标准消化和洗涤剂辅助消化富含的蛋白质组,以对 SP3 与 SP3 (DA) 和 SP4 与 SP4 (DA) 进行成对比较。通过全细胞裂解物(图 6)和膜组分(图 S9)中的标准消化富集的蛋白质组映射到细胞质、膜、核和细胞骨架细胞成分本体。而由去污剂辅助消化富集的蛋白质组仅映射到膜细胞成分本体(图 6 和 S9)。这些结果进一步证实,与标准消化相比,去污剂辅助消化提高了检测膜蛋白的能力。
SP3 与 SP4 的蛋白质捕获和富集的机制差异
SP3 最初被提议通过类似 HILIC 的机制捕获蛋白质,使用两种类型的亲水性、羧酸盐修饰的颗粒,这些颗粒的表面亲水性不同。(11) 然而,最近的一项研究表明,在 SP3 中,蛋白质聚集主要负责蛋白质捕获,而不是 HILIC 样相互作用驱动 SP3 中的蛋白质捕获。如果 SP3 的蛋白质捕获仅依赖于蛋白质聚集,我们预计 SP3 和 SP4 中鉴定的蛋白质会产生相似的蛋白质鉴定,尤其是在检查样品复杂性降低的亚细胞组分时。然而,我们的结果表明,每种方法鉴定的蛋白质组存在明显差异(图 2),SP3 和 SP4 鉴定的单个组分中不到 45% 的蛋白质组。我们的结果表明,在全细胞裂解物和膜组分中,SP4 能够鉴定和富集与 SP3 鉴定的蛋白质相似或更疏水的蛋白质。疏水性蛋白质含有更多的不溶性氨基酸,这使得蛋白质在蛋白质聚集机制中添加有机溶剂后能够很好地聚集。这些结果与之前的工作一致,后者表明与 SP3 相比,SP4 在细胞裂解物中富集了低溶解度蛋白质。
我们的结果还表明,对于预期含有亲水性蛋白质的组分,SP3 能够识别(图 1-3)和富集(图 4-6)与 SP4 相比相似或更亲水性的蛋白质。如果蛋白质聚集驱动了 SP3 中的蛋白质捕获,那么与我们观察到的 SP4 相比,我们预计会看到具有相似富集水平的相似蛋白质。然而,与 SP4 相比,我们观察到 SP3 鉴定出的亲水性蛋白更多,这一事实表明,与羧酸盐修饰颗粒的 类HILIC 相互作用也在捕获 SP3 蛋白中发挥作用。亲水性蛋白质对极性官能团(如羧酸盐)具有更大的亲和力,这种亲和力基于氨基酸序列中极性和带电官能团的数量,这些官能团可以通过氢键、偶极相互作用和盐桥与羧酸盐基团相互作用。蛋白质的氨基酸序列越亲水,就越有可能与羧酸盐修饰的颗粒相互作用。因此,这些结果表明 SP3 和 SP4 的蛋白质捕获机制不同,每种纯化技术的蛋白质捕获都取决于蛋白质疏水性。
结论
蛋白质组学实验能够鉴定生物学相关样品中的肽和蛋白质,以确定疾病中的重要蛋白质生物标志物。在此,我们比较了 SP3、SP4、SP3(DA) 和 SP4(DA),并表明 SP3 和 SP4 蛋白质捕获机制与每种纯化技术的蛋白质捕获不同,具体取决于蛋白质疏水性。我们的结果表明,SP3 通过类 HILIC相互作用和蛋白质聚集机制的组合捕获蛋白质,而 SP4 似乎仅通过蛋白质聚集机制捕获蛋白质。此外,去污剂辅助消化改善了全细胞裂解物和膜组分中疏水性膜蛋白的鉴定。SP3磁珠请参考http://www.purimagbead.com/Product/1736805216.html。
尽管理想的蛋白质组学方法可以成功捕获生物样品中的所有蛋白质,但我们表明 SP3 和 SP4 捕获不同的蛋白质。因此,样品纯化技术的选择(SP3 与 SP4)可能会使蛋白质组学实验中鉴定的蛋白质产生偏差。为了捕获具有不同疏水性的多种蛋白质,可能需要多种样品净化技术。或者,如果研究人员有兴趣鉴定具有预期疏水性的目标一类蛋白质,例如预期具有疏水性的跨膜蛋白,则可以选择特定的样品净化方法,例如 SP4。最终,选择最佳样品纯化方法可以增加复杂样品的蛋白质组覆盖率,这将有助于深入分析蛋白质在调节细胞活性和导致疾病方面的重要作用。
- 上一篇:使用 SP3 纯化基于凝胶的样品分级用于自上而下的蛋白质组学 2025/4/15
- 下一篇:视角:去中心化的纳米颗粒蛋白冠分析可能误导生物标志物的发现 2025/4/5