
基于纳米颗粒的血浆蛋白质组学中血液成分污染的系统性评估
无独有偶,近日,德国马普所的蛋白组学大神Matthias Mann,和他的知名高足Phillipp Geyer在bioarxiv上发表一篇题为“Pre-Analytical Drivers of Bias in Bead-Enriched Plasma Proteomics”的研究结果,剑指在质谱血浆蛋白组学领域一直存在的蛋白鉴定数据质量良莠不齐、缺乏标准,甚至人为数据造假的乱象。参考我站评述文章http://www.purimagbead.com/html/1086233051.html,与本篇解读遥相呼应。
研究团队通过系统优化纳米颗粒富集血浆蛋白质的条件,建立了一个高灵敏度的流程OmniProt,构建了覆盖超过1万个血浆蛋白的质谱图库,并开发了自动化污染识别软件Baize,全面提升了血浆蛋白质组学的鉴定深度和实验稳定性与临床可用性。

图1 文章截图
本研究系统评估了血小板、红细胞和凝血相关污染对基于纳米颗粒(nanoparticle, NP)的血浆蛋白质组学实验的影响,并初步探讨了凝血因素的干扰,发现血小板和红细胞污染是导致血浆样本蛋白鉴定数量升高和定量结果变异的重要因素之一。研究团队开发了基于二氧化硅纳米颗粒的高通量血浆蛋白质组流程 OmniProt,建立了覆盖10,109个蛋白组的高质量谱图库,并通过污染模型分别筛选出三类污染的特异性蛋白标志物,最终构建了开放获取的软件工具Baize用于污染评估。该方法在199例肺结节临床队列中得到验证,表现出高通量、高灵敏度和良好的数据准确性,为血浆蛋白质组在疾病生物标志物研究和临床转化中的可靠性与实用性提供了有力支持。尽管纳米颗粒富集技术近年来被广泛应用于提升质谱在血浆中的蛋白鉴定深度,但不同研究之间蛋白鉴定数量差异显著(2000至7000个不等),引发对其稳定性与可靠性的质疑。尤其是血小板、红细胞或凝血成分的污染,作为临床采样中常见却易被忽视的问题,可能是造成结果波动的重要原因。为此,本研究通过开发高效的NP富集流程OmniProt、建立大规模血浆谱库,并引入Baize软件系统性评估污染影响,全面揭示了污染在血浆蛋白质组分析中的干扰机制,并验证了该策略在肺癌临床样本中的可行性与诊断潜力。

图2 研究流程
01. 纳米颗粒的筛选与性能评估
研究首先针对低丰度血浆蛋白的富集需求,系统比较了不同类型的二氧化硅纳米颗粒,包括固体球形、介孔、空心介孔及分级孔结构等类型。
在肺癌患者血浆样本的重复分析中,所有纳米颗粒之间鉴定的蛋白质重叠度超过80%,且实验重复性良好(CV为1–2%),表明该纳米颗粒富集策略技术稳定性高。结果显示,固体二氧化硅纳米颗粒在蛋白质(protein groups)与肽段(peptides)的鉴定数量上均优于其他类型,分别提高了约16%和33%。
进一步比较不同直径的固体颗粒(300–1000 nm)后发现,尽管蛋白鉴定数目差异不大,但从表面积、离心洗涤效率及操作便捷性等因素综合考虑,500 nm直径颗粒(NP23)在性能与实用性之间达成了良好平衡,因此被选为后续工作的最佳纳米颗粒材料。

图3 不同类型的二氧化硅纳米颗粒
02. OmniProt血浆蛋白质组流程的建立
在确立NP23为富集载体后,研究优化了关键步骤,包括血浆稀释、蛋白冠形成、洗脱软冠以及纳米颗粒的储存条件。在稀释条件对比中,碱性缓冲液(pH 11)显著提高了蛋白与肽段的鉴定数量(分别提升12%与36%),并在扫描电镜观察中显示出稳定的纳米颗粒结构与蛋白冠形成。在优化颗粒用量和孵育时间的实验中,发现使用0.5 mg纳米颗粒、30分钟孵育为最佳条件。三轮洗涤及7000 g离心条件被证明是有效去除软蛋白冠的方式。此外,短期在4–30°C范围内储存不影响其蛋白富集性能,表明该系统具备较好的温度稳定性。这些系统性的优化奠定了OmniProt流程的标准化基础,确保了其在血浆蛋白质组学中的稳定性与重复性。
03. 构建10,000+蛋白质组的血浆谱图库
为支持后续数据非依赖性采集(DIA)分析,研究团队利用21种化学特性不同的纳米颗粒对20位肺癌患者的血浆样本进行蛋白质富集,并通过高pH反相色谱将肽段分为30或60个组分,进而进行Astral nDIA分析,总计生成780个DIA数据文件。
通过DIA-NN分析,最终建立了包含126,661个肽段离子、对应10,109个蛋白组的高覆盖度谱库。该数据库覆盖了人类血浆蛋白质图谱中94%的已知蛋白,并显示出对多种NP特性(如电荷、官能团、亲水/疏水性)的响应差异。厦门普睿迈格提供各种不同特性的磁珠(http://www.purimagbead.com/Product/MSpretreatment/),可用于评估其在血液样本中的蛋白冠形成情况。
功能富集分析表明,这些蛋白涉及信号转导、凋亡、蛋白磷酸化、细胞粘附及先天免疫等多个生物过程,同时揭示了该资源对神经退行性疾病、代谢通路及肿瘤通路的显著覆盖能力,体现其广泛的应用潜力。
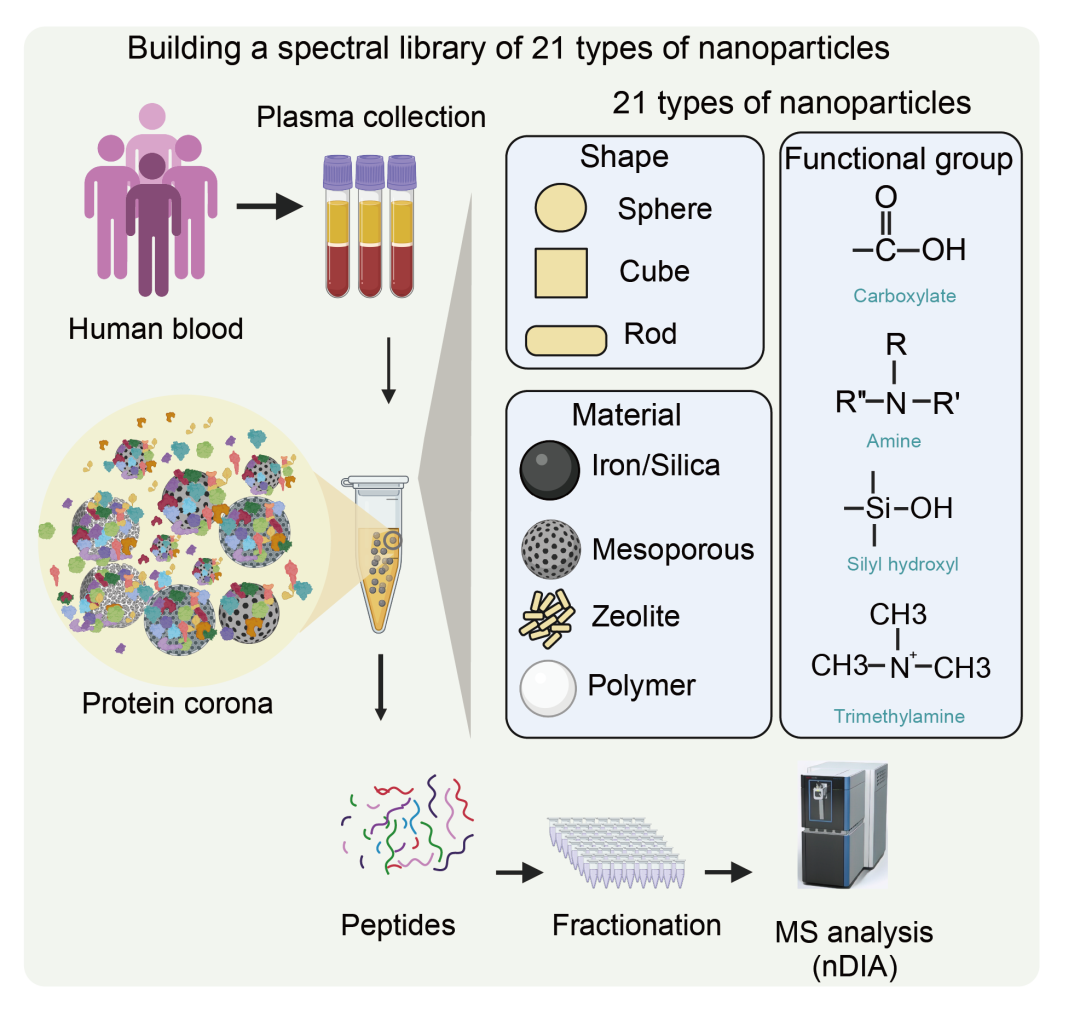
利用21种化学特性不同的纳米颗粒对20位肺癌患者的血浆样本进行蛋白质富集
04. OmniProt对低丰度蛋白的检测性能评估
研究将OmniProt与商用Top14高丰度蛋白去除试剂盒及传统直接酶解(Neat)方法进行比较。结果表明,OmniProt在蛋白组与肽段鉴定数量上分别比Top14高1.7倍和2.6倍,比Neat方法高4.8倍和5.7倍,显著提升了低丰度蛋白的检测能力。尤其在log10强度为3.2–4.5的区间内,OmniProt能检测更多低丰度蛋白。此外,在人-牛混合样本的交叉物种定量精度测试中,OmniProt在检测蛋白数量与定量准确性方面均优于直接分析组,且生物学重复之间的变异系数中位数低于20%,证实了该流程的高灵敏度与高重复性,适用于复杂背景中的精准蛋白定量分析。

图5 OmniProt的定性和定量评估
05. 不同血浆样本中蛋白鉴定数量的变异性
在对OmniProt流程优化中发现,在不同的血浆样本应用中观察到显著的蛋白鉴定数量差异,范围从约3000至7000个蛋白组不等。
进一步分析表明,蛋白鉴定数目较高的样本中存在大量血小板或红细胞来源的蛋白,提示样本可能受到这些细胞成分的污染。这一发现引发了研究团队对血液污染对蛋白质组分析干扰机制的深入探讨,并为后续污染标志物的鉴定与评估工具的开发提供了研究动因。
06. 血小板污染及其标志物发现
为了系统评估血小板污染对NP富集流程的影响,研究构建了PRP(富血小板血浆)与PPP(贫血小板血浆)的混合梯度样本,并使用Neat和OmniProt流程进行处理。
结果表明,血小板污染显著增加蛋白鉴定数量,同时干扰低丰度循环蛋白的定量准确性。现有文献的30个血小板污染标志物在OmniProt处理下表现相关性降低,提示需建立适用于基于纳米颗粒流程的新标志物集。通过Mfuzz聚类与相关性分析,筛选出30个与血小板污染程度高度相关的新标志物(Spearman r > 0.92),涵盖血小板功能相关蛋白,并通过12例患者样本验证。
研究还开发了Baize软件,通过这些标志物实现污染程度评估(R² = 0.95)。此外,在不同纳米材料处理下,该标志物集表现稳定,具备跨平台适用性,但污染指数的评估不可在跨纳米材料直接比较。
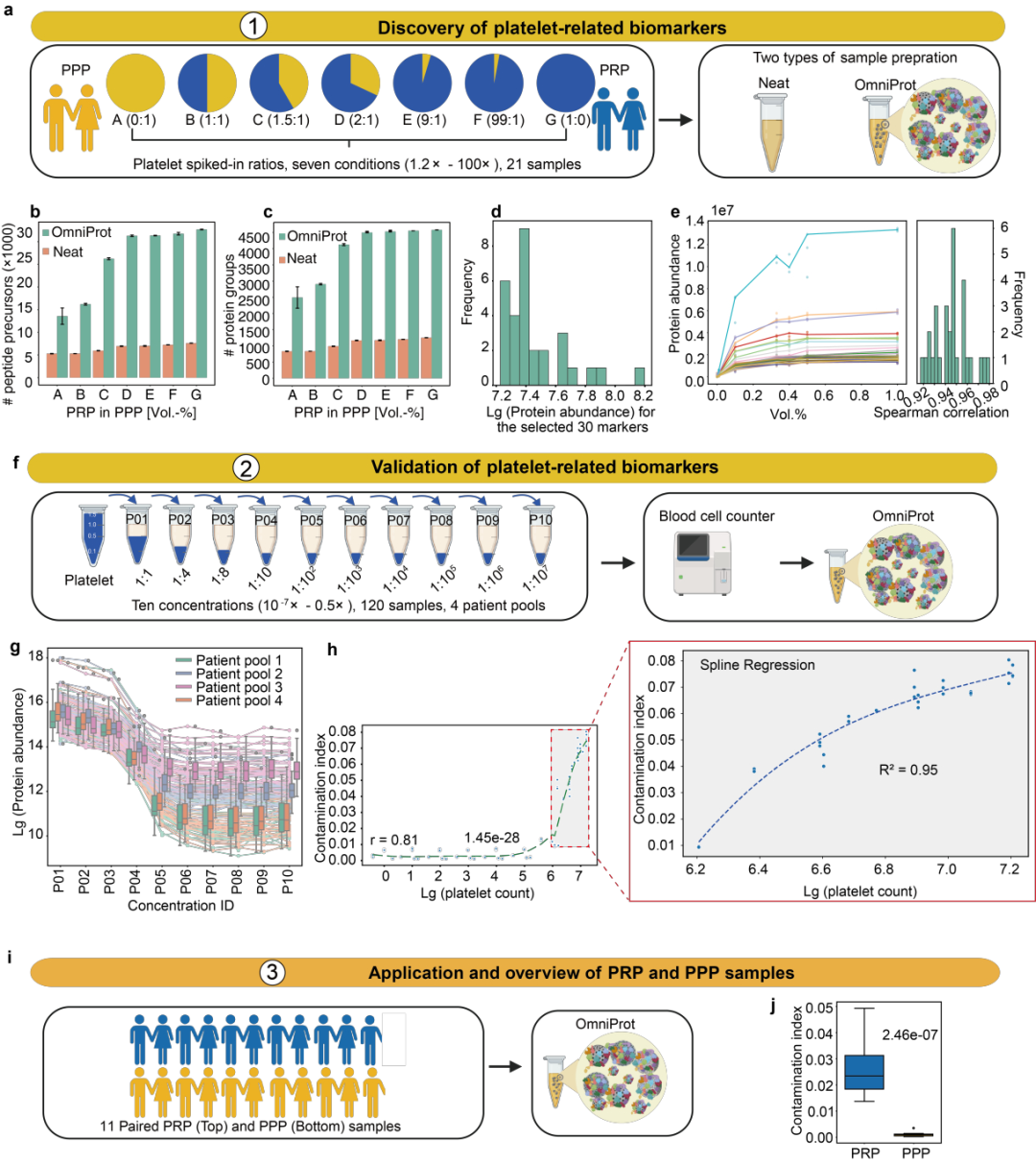
图6 用于评估血小板污染的生物标志物的发现、验证及应用
07. 红细胞污染及其标志物发现
为研究红细胞污染对NP蛋白质组的影响,研究人员设计了红细胞-PPP混合梯度模型。红细胞污染同样导致蛋白识别数量增加,并干扰红细胞无关蛋白的定量。研究从高相关性(r>0.98)蛋白中筛选出30个与红细胞污染相关的新标志物,涵盖血红蛋白、细胞膜骨架及气体转运蛋白,具有明确生物学特异性。通过六例患者的稀释梯度实验进一步验证,并开发红细胞污染指数,集成入Baize软件,实现准确定量(R²=0.94)。
08. 凝血相关的污染及其标志物的发现
研究指出,血浆样本在采集过程中如操作不当,易因凝血不全而形成血浆-血清混合状态,影响蛋白质组学的定量结果。
为评估抗凝剂的影响,研究人员比较了使用三种不同抗凝管(EDTA、柠檬酸钠、肝素)采集的样本,经Neat与OmniProt处理后的质谱结果。蛋白识别总量无明显差异,但仅59.2%的蛋白在三种抗凝剂中同时被检测到,说明不同类型的采血管对蛋白谱具有影响。功能分析显示,OmniProt在B细胞发育、翻译起始、免疫及病毒复制相关蛋白上有更强富集。
随后研究比较了血浆样本(紫头管)与血清样本(红头管),经OmniProt处理后,血浆和血清中蛋白识别数量均显著高于Neat方法(3041 vs 816;3066 vs 796)。通过筛选上下调显著(fold change ≥ 5,p < 0.05)的蛋白,确定了20个与凝血相关的污染标志物(如FGA、FGG、FGB、AGGF1、TFRC),并将其整合为凝血污染评估模型,引入Baize软件。
09. Baize软件:NP血浆污染评估工具
研究人员开发了名为 Baize的网页工具,用于快速评估NP富集血浆样本中的三类关键污染:血小板、红细胞和凝血相关污染。
软件通过计算每类污染标志物信号强度与总蛋白信号之比,生成污染指数(Contamination Index)。用户上传蛋白表达矩阵后,Baize可自动输出各样本的污染状态。该工具免费开放,地址为:https://www.guomics.com/Baize
10. OmniProt和Baize在肺癌临床队列中的应用
为验证OmniProt和Baize的实际应用价值,研究分析了199例肺结节患者(含良性、早期恶性及6例晚期对照)的血浆样本。样本采用OmniProt流程处理后进行Astral 质谱仪的nDIA方法采集数据,平均每例鉴定出约4413个蛋白组,实验重复性高。通过Baize检测出5例存在明显污染,已从后续分析中剔除。
随后,研究人员采用8种机器学习算法对良恶性结节进行分类,发现Extra Trees模型在F1分数、准确率、召回率及AUC方面表现最优。
模型最终筛选出与肺癌相关的关键蛋白如FSCN1、PDGF、FCGR3A、HAMP、COL15A1等,表明该蛋白组数据具备区分肺癌病理状态的潜力,尽管进一步验证仍需独立队列支持。
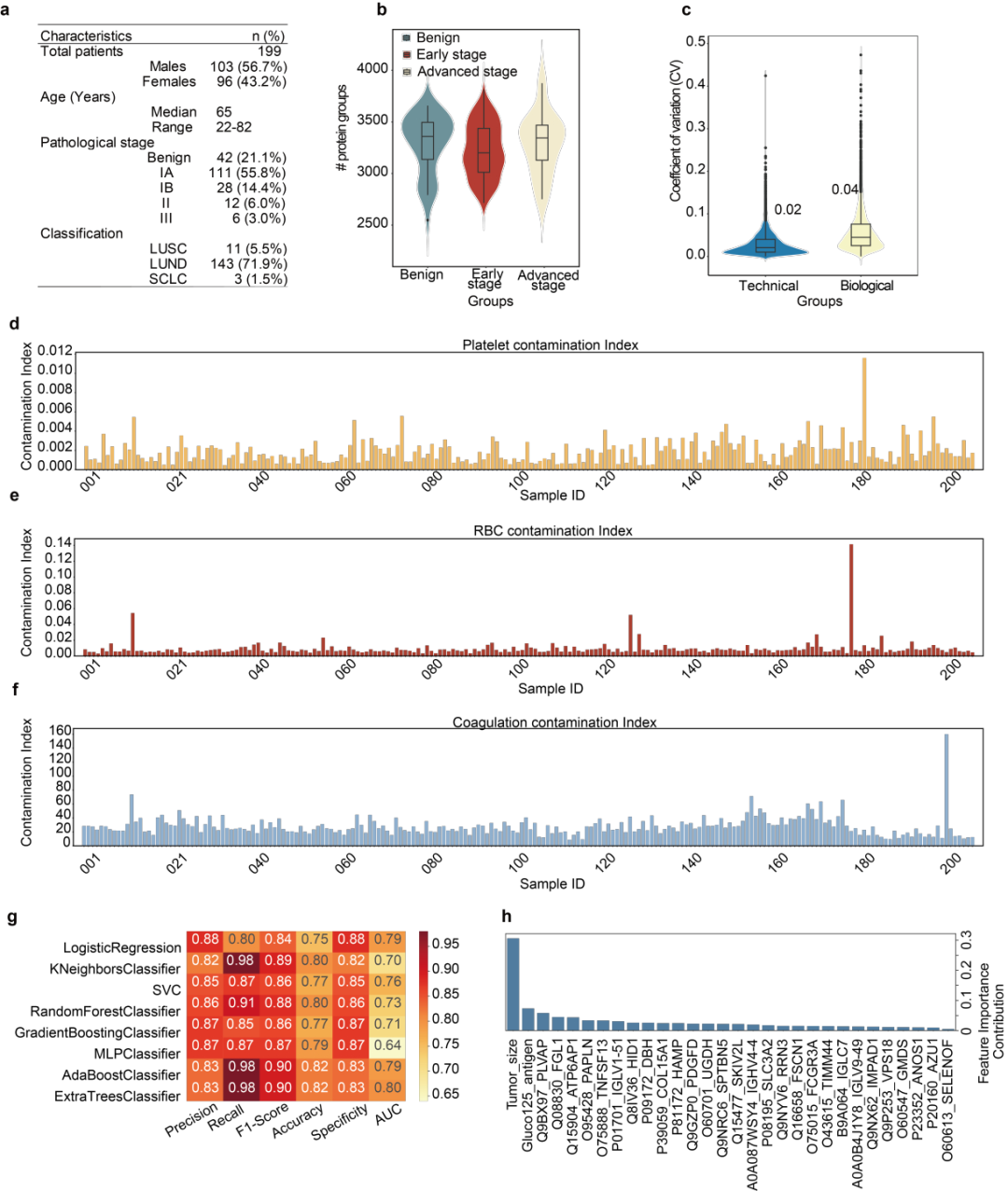
图7 OmniProt在肺癌临床队列中的应用
研究首次系统阐明了血液污染对基于纳米颗粒富集血浆蛋白质组学实验的广泛影响,提供了技术流程(OmniProt)、计算工具(Baize)与数据资源(谱库)三位一体的解决方案,为血浆蛋白质组的高通量精准测定及其在临床疾病生物标志物研究中的应用奠定了坚实基础。
- 上一篇:蛋白互作检测之磁珠法Co-IP技术 2025/5/18
- 下一篇:Mann神出手警惕血浆蛋白组学数据中可能的污染!!血浆蛋白组 2025/5/17